
文献解读 | 高密度脂蛋白通过miR-181a-5p调控自噬影响血管新生
正常的高密度脂蛋白(nHDL)在防治组织缺血性疾病中起到积极作用,促进血管生成。相反,在冠心病等病理情况下,高密度脂蛋白(HDL)的功能可能受损,失去促进血管生成的效果,即失功能HDL(dHDL)。自噬是维持细胞和生物体稳态的重要机制。微小RNA(miRNA)在调节血管生成中发挥关键作用。目前尚不清楚HDL是否通过miRNA调节自噬来影响血管新生,以及dHDL是否在调节自噬方面与nHDL有所不同。
近日,中山大学附属第一医院区景松和欧志君课题组在Science China Life Sciences发表了题为“High-density lipoprotein regulates angiogenesisby affecting autophagy via miRNA-181a-5p”的研究论文,揭示了HDL通过miR-181a-5p调控自噬流影响血管新生的新机制。

nHDL通过刺激自噬和eNOS表达增加一氧化氮(NO)生成,从而促进血管生成。相反,dHDL通过增加miR-181a-5p表达降低自噬和eNOS表达,导致NO产生减少,最终抑制血管生成。这一发现提供了高密度脂蛋白调节血管生成的新机制,为治疗dHDL妨碍的血管生成提供了治疗靶点。
技术路线
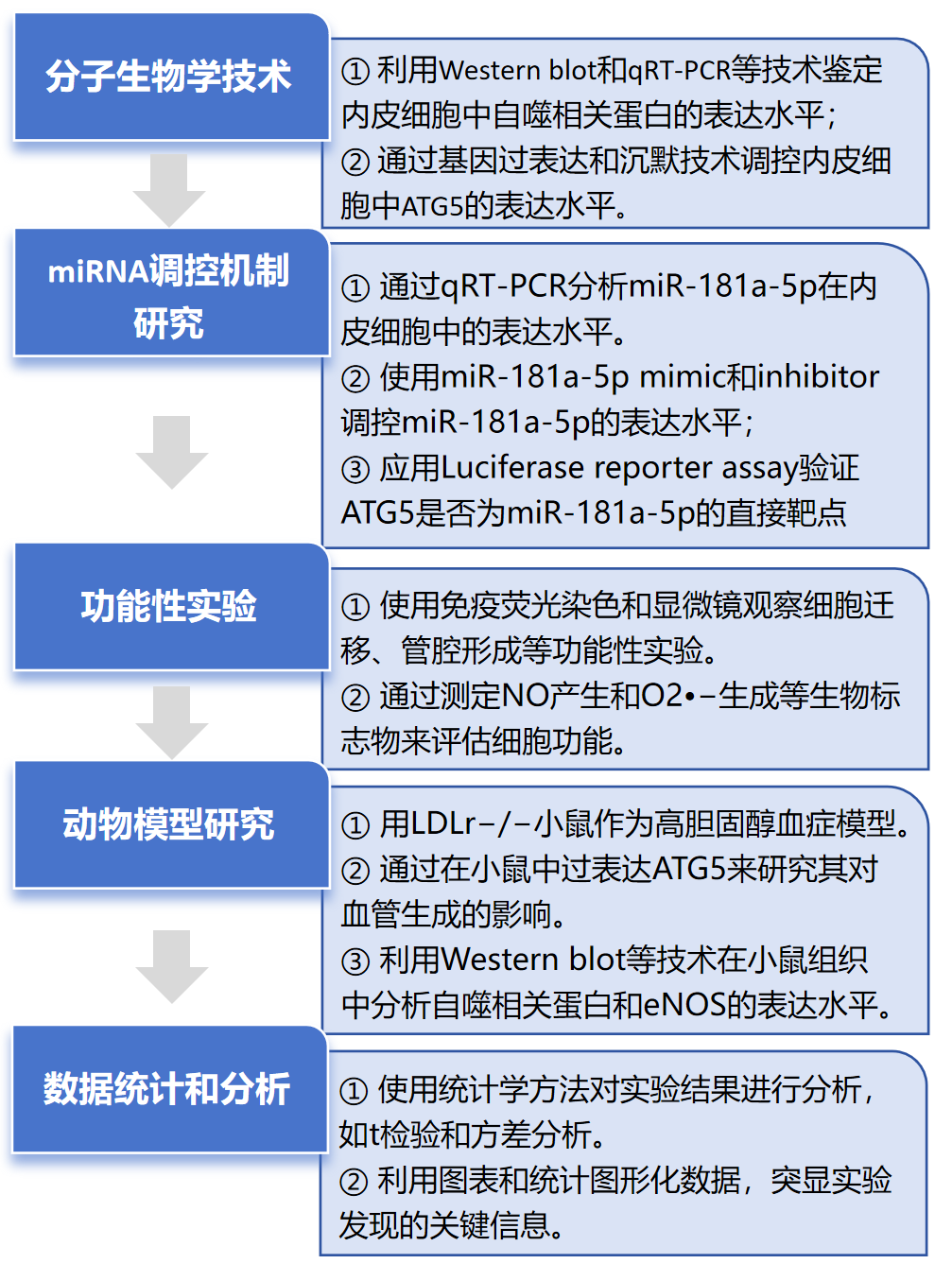
其中Luciferase reporter assay由金开瑞生物合作完成
研究方法及结果
nHDL促进HUVECs中的自噬,而dHDL抑制
研究使用了人脐静脉内皮细胞(HUVECs)来调查正常高密度脂蛋白(nHDL)和冠心病患者高密度脂蛋白(dHDL)对自噬的影响。通过测量自噬相关蛋白和使用荧光探针等方法,研究发现,与nHDL相比,dHDL显著增加了细胞内SQSTM1/P62水平,减少了LC3-II的表达。此外,nHDL促进了ATG5、Beclin1和PI3K-CIII等自噬相关蛋白的表达,而dHDL则产生相反的效应。通过使用荧光标记系统,研究还观察到nHDL增加了细胞内自噬体和自溶体的形成,而dHDL则抑制了这一过程。这些结果表明,nHDL和dHDL对HUVECs中的自噬产生不同的调节作用。
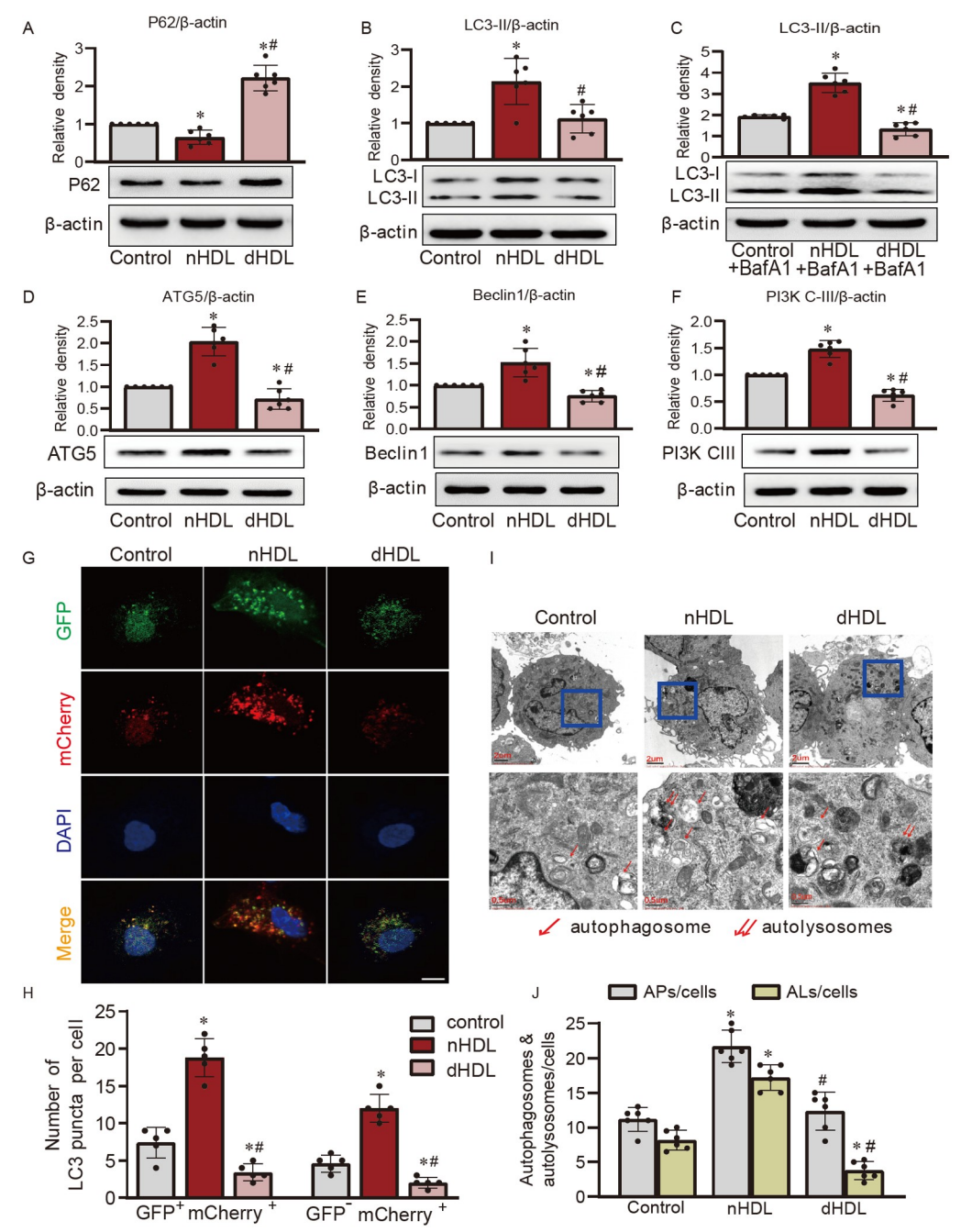
图1. nHDL和dHDL对内皮细胞自噬的影响。
A–C,Western blot和定量分析显示在nHDL或dHDL刺激下,HUVECs中LC3-II和P62的水平;D–F,在nHDL或dHDL刺激下,通过Western blot检测HUVECs中ATG5、Beclin1和PI3K-CIII的表达,;G,通过共聚焦图像显示nHDL和dHDL对表达自噬通路报告基因mCherry-GFP-LC3的HUVECs中自噬的影响;H,对在合并图像中测得的自噬体(GFP+mCherry+)和自溶体(GFP−mCherry+)的定量分析;I和J,电镜图像和定量分析显示在nHDL或dHDL刺激下HUVECs中的自噬体或自溶体。
在人脐静脉内皮细胞(HUVECs)中通过抑制自噬降低eNOS和NO的产生
通过使用3-甲基腺嘌呤(3-MA)抑制自噬,验证了nHDL和dHDL诱导的自噬在内皮细胞(ECs)中的不同功能。图2A和B显示,当抑制自噬时,nHDL导致的PI3K-CIII和eNOS的上调以及P62的下调被抑制。抑制自噬时,nHDL诱导的NO产生受阻(图2C–E)。自噬抑制增加了O2•−的产生(图2F和G)。这些数据为我们了解nHDL如何通过自噬途径保护ECs免受氧化应激提供了新的见解。
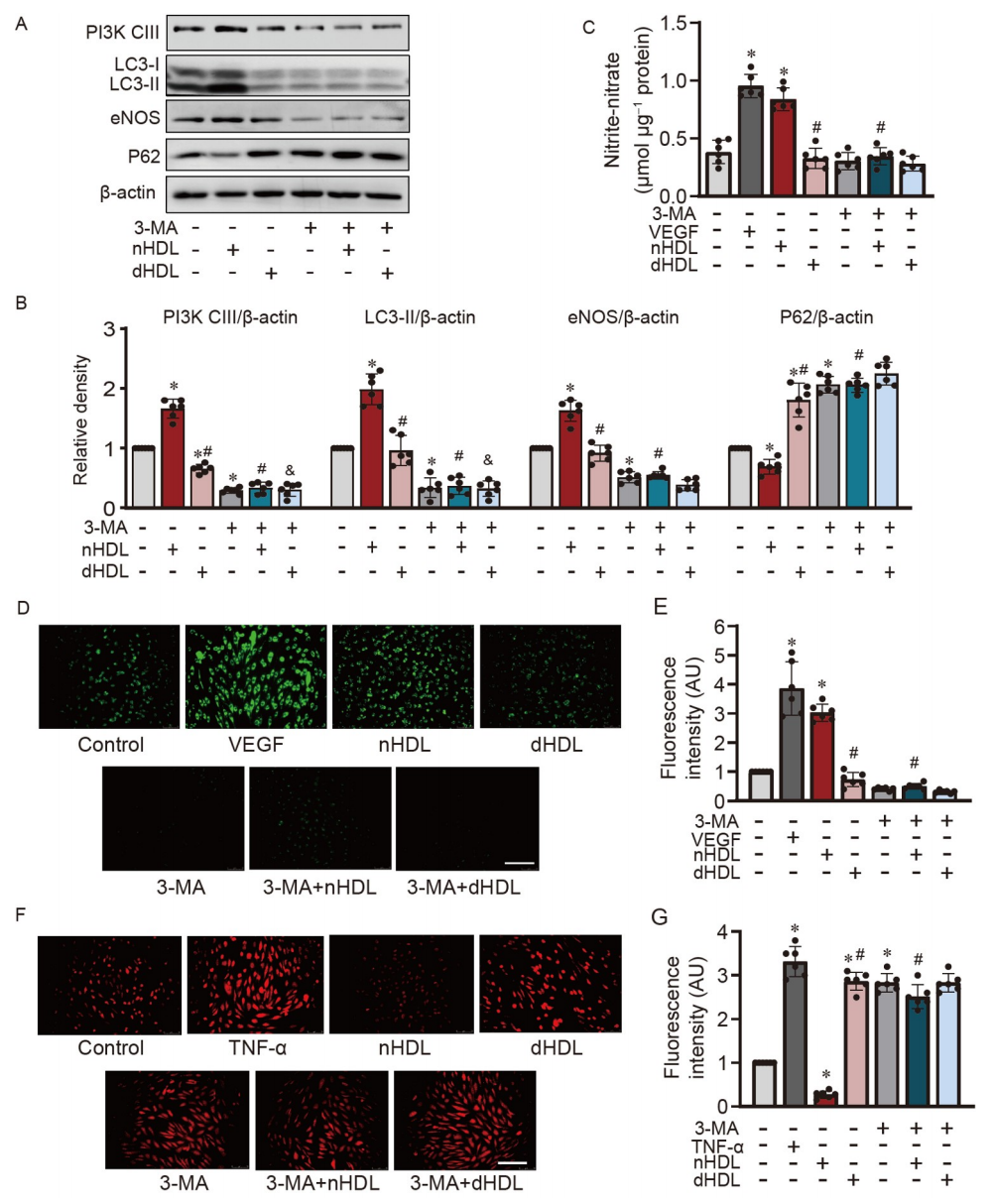
图2. 抑制自噬对nHDL促进HUVECs中eNOS表达、NO产生和O2•−生成的影响。
A,Western blot检测经nHDL或dHDL处理的HUVECs中LC3-I/II、eNOS、PI3K-CIII和P62的表达;B,蛋白定量相对于beta-actin表达;C,使用Sievers NOA分析仪显示HUVECs中的NO产生的条形图;D和E,使用DAF-2DA检测在HUVECs中经nHDL或dHDL处理后细胞内NO的图像和定量;F和G,在HUVECs中,图像和定量显示3-MA通过抑制自噬增强了对照组和nHDL处理组中内皮O2•−的生成。
通过沉默ATG5抑制自噬导致内皮细胞中O2•−的增加,并减弱了nHDL诱导的内皮细胞迁移和血管形成
ATG5在自噬体形成中至关重要,对于细胞分化和稳态维持起着重要作用。由于nHDL和dHDL对ATG5有显著不同的影响(图1D),作者选择ATG5作为自噬干预的靶点。在HDL处理之前沉默了ATG5的表达。ATG5的沉默效果显示在支持信息的图S2中。ATG5的沉默降低了LC3-II和eNOS的表达(图3A和B),增加了P62的表达,阻断了nHDL诱导的eNOS表达和NO产生(图3C和G),增强了O2•−的生成(图3D和H)。此外,沉默ATG5抑制了血管内皮细胞nHDL诱导的细胞迁移(图3E和I)和管形成(图3F和J)。
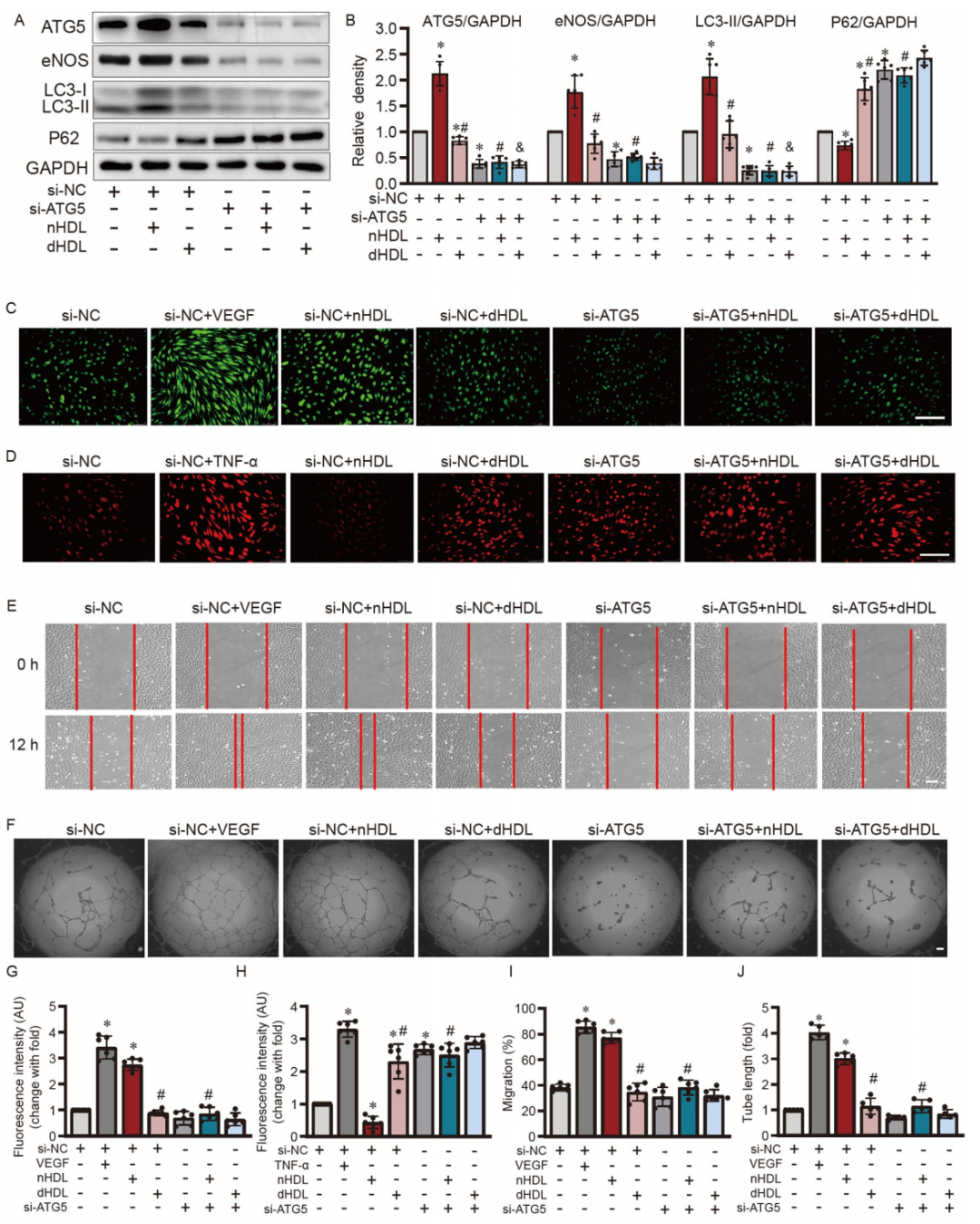
图3. ATG5沉默和nHDL诱导的eNOS表达、NO产生、细胞迁移、管形成、以及在HUVECs中O2•−生成的定量分析。
A和B,Western blot和定量分析显示ATG5的沉默降低了eNOS和LC3-II的表达,同时增加了P62在培养的HUVECs中的表达;C和D,图像显示当通过ATG5沉默减弱自噬时,nHDL或dHDL对NO产生和O2•−生成的影响;E和F,图像显示siRNA沉默ATG5抑制了nHDL诱导的HUVECs中的迁移和血管形成;G–J,定量分析显示siRNA沉默ATG5抑制了nHDL诱导的HUVECs中的NO产生、迁移、血管形成,以及O2•−生成。
通过过表达ATG5来刺激自噬,挽救了dHDL的血管生成作用
为了探究刺激自噬是否能够逆转dHDL的血管生成效应,研究者在内皮细胞中过表达了ATG5。ATG5的过表达增加了LC3-II和eNOS的表达,降低了在dHDL刺激下的P62的表达。ATG5的过表达恢复了由dHDL抑制的NO产生,减少了dHDL刺激下的O2•−生成。重要的是,ATG5的过表达在dHDL刺激下恢复了内皮细胞的迁移和管道形成。这些数据表明ATG5可能是nHDL保护作用的介导者,并对内皮细胞的血管生成产生了显著影响。
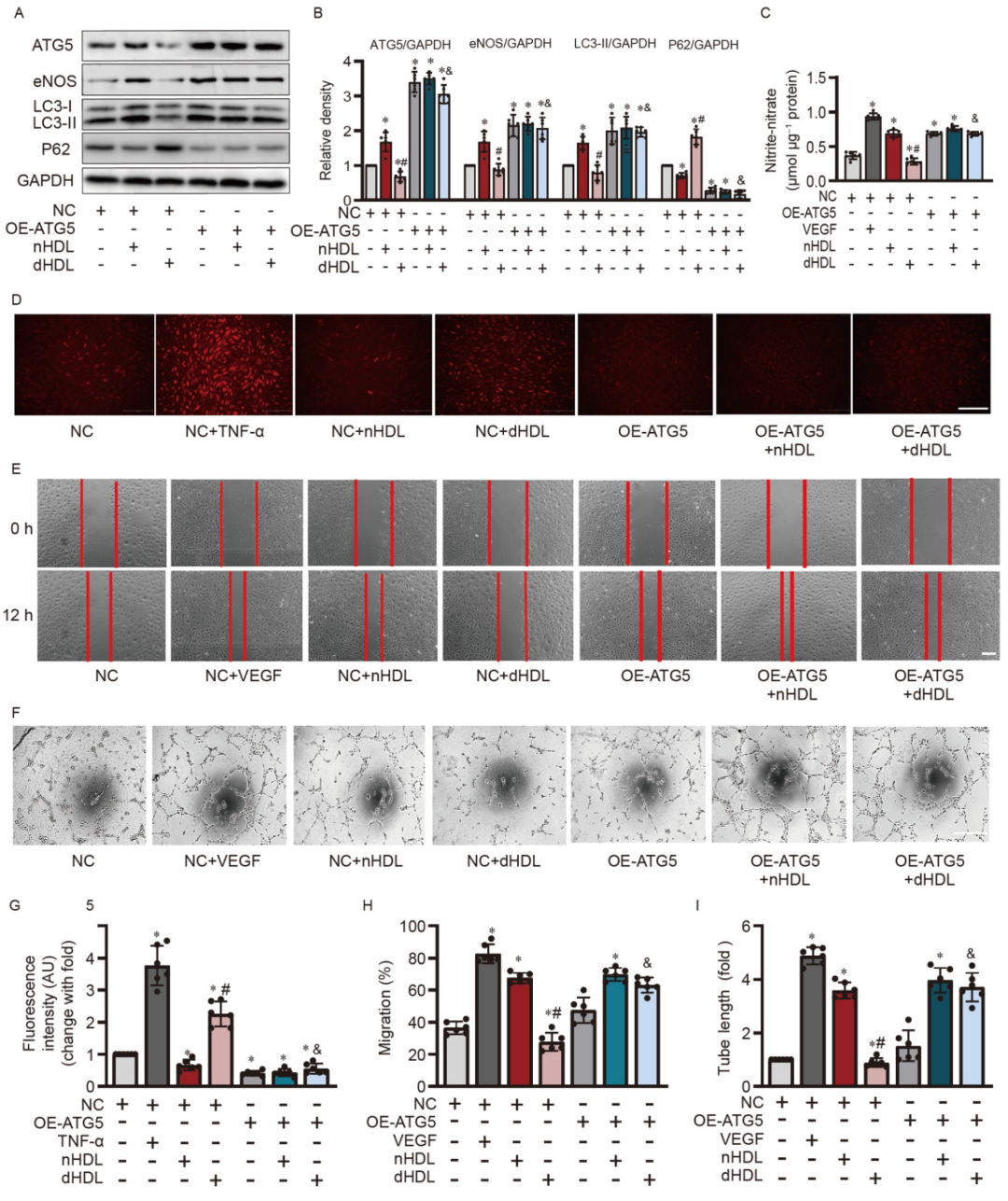
图 4. ATG5的过表达恢复了dHDL抑制的HUVECs中eNOS的表达,NO的产生,迁移和管道形成,并降低了O2•−的生成。
A和B,Western blot和定量分析显示ATG5的过表达增加了eNOS和LC3-II的表达,并抑制了P62在培养的HUVECs中的表达;C,检测了在过表达ATG5后,nHDL或dHDL处理下HUVECs中的内皮NO产生,NO的产生增加,并恢复了dHDL促进HUVECs中NO产生的能力;D,图像显示了ATG5的过表达减少了dHDL诱导的HUVECs中O2•−的生成;E和F,图像显示了ATG5的过表达,恢复了HUVECs中dHDL诱导的迁移和血管生成。G–I,对D–F图像的定量分析。
nHDL和dHDL通过miR-181a-5p以不同方式调控ATG5
研究者重新验证了数据(Li等,2020),证实miR-181a-5p在nHDL作用下降低而在dHDL作用下上升,通过定量实时聚合酶链反应(qRT-PCR)进行检测(图5A)进一步发现nHDL抑制了pre-miR-181-1和pre-miR-181-2,而dHDL刺激了它们(图5B)。在沉默SRB1后,nHDL和dHDL对miR-181a-5p的影响被消除(图5C),表明它们通过SRB1受体在ECs中调节miR-181a-5p的表达。TargetScan(targetscan.org)显示ATG5可能是miR-181a-5p在HUVECs中的潜在靶点(图5D)。miR-181a-5p模拟物抑制了ATG5的Luciferase活性,验证了ATG5是miR-181a-5p的直接靶标(图5E)。将miR-181a-5p模拟物和抑制物转染到HUVECs后,miR-181a-5p模拟物降低了ATG5的表达,而miR-181a-5p抑制物增加了它们(图5F–H),同时eNOS的表达也受到了类似的调控(图5G和H)。
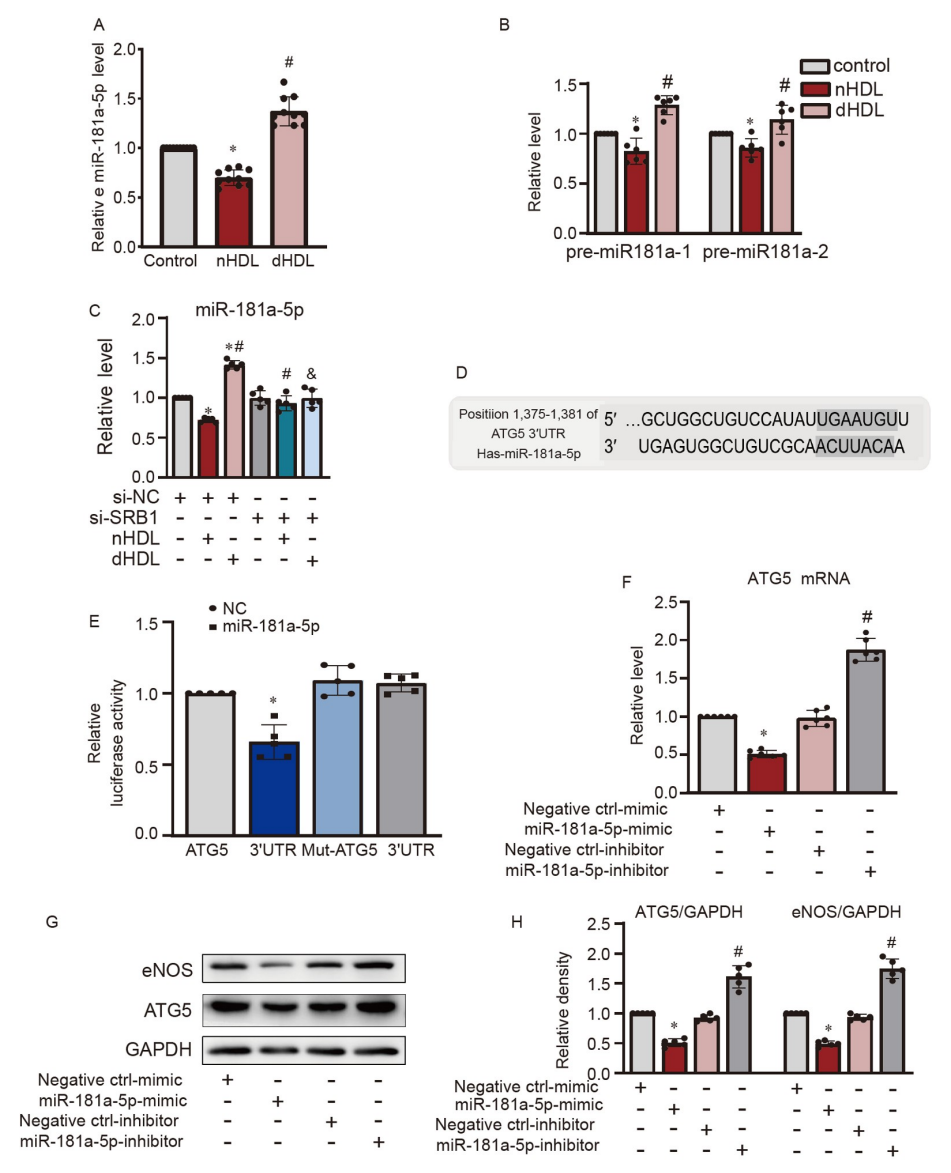
图5. HDL对内皮miR-181a-5p和ATG5表达的影响
A,qRT-PCR显示nHDL和dHDL对培养的HUVECs中miR-181a-5p表达的影响;B,qRT-PCR显示nHDL和dHDL对培养的HUVECs中pre-miR-181-1和pre-miR-181-2表达的影响;C,qRT-PCR显示在沉默SRB1后,nHDL和dHDL对培养的HUVECs中miR-181a-5p表达的影响;D,ATG5中可能的人类miR-181a-5p结合位点;E,Luciferase活性确认ATG5在HUVECs中是miR-181a-5p的直接靶标;F,miR-181a-5p模拟物降低了HUVECs中ATG5的mRNA表达,而miR-181a-5p抑制物则增加了它;G和H,miR-181a-5p模拟物或miR-181a-5p抑制物转染到HUVECs中后,eNOS和ATG5的免疫印迹分析的代表图像和定量分析。
研究者进一步研究了HDL是否通过miR-181a-5p调节自噬。miR-181a-5p模拟物抑制了LC3-II的表达,增加了P62的表达,阻断了自噬体的合成(图6A、C、E和F)。miR-181a-5p抑制物增加了LC3-II的表达,抑制了P62,促进了自噬体的合成(图6B、D、E和G)。miR-181a-5p模拟物抑制了nHDL诱导的自噬,miR-181a-5p抑制物逆转了dHDL抑制的自噬(图6A–G),证明miR-181a-5p介导了HDL对自噬的调节。
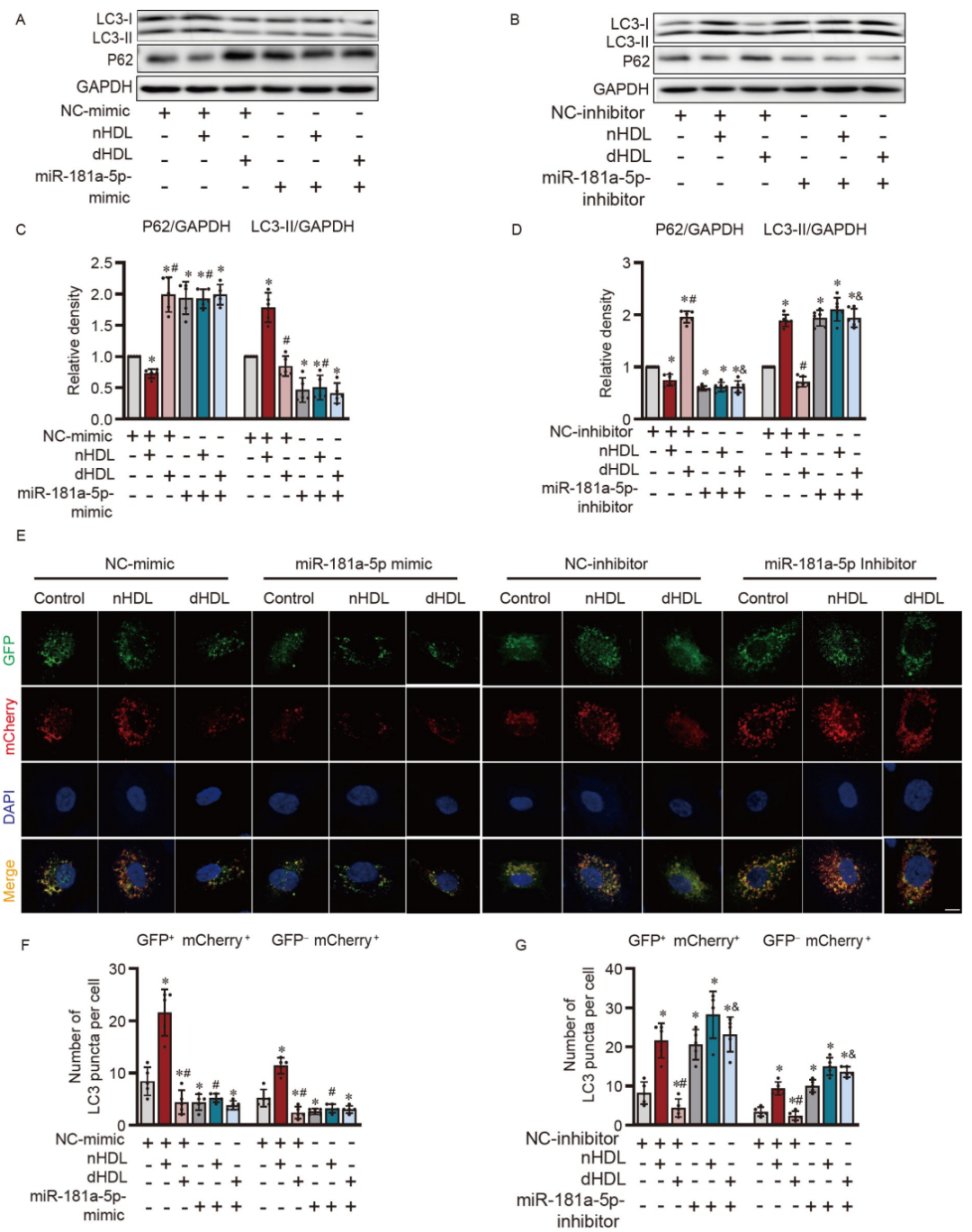
图6. HDL对内皮自噬和miR-181a-5p调控的影响。
A和B,WB显示转染miR-181a-5p模拟物或抑制物的HUVECs在nHDL或dHDL刺激后LC3-I/II和P62的表达;C和D,A和B中图像的定量分析;E,共聚焦图像显示在转染miR-181a-5p模拟物或抑制物后,通过在HUVECs中使用自噬通路报告基因mCherry-GFP-LC3来观察nHDL和dHDL对自噬的影响;F和G,合并图像中测量的自噬体(GFP+mCherry+)和自溶体(GFP−mCherry+)的定量。
nHDL和dHDL通过miR-181a-5p不同程度地影响内皮细胞的NO产生、O2•−生成、迁移和血管形成
研究者进一步研究了HDL是否能通过miR-181a-5p来调节内皮功能。图7A–D显示,miR-181a-5p模拟物抑制了nHDL诱导的内皮细胞中的NO产生,而miR-181a-5p抑制剂逆转了dHDL抑制的NO产生。miR-181a-5p模拟物增加了nHDL处理的内皮细胞中的O2•−生成(图7E和G),而miR-181a-5p抑制剂降低了dHDL诱导的O2•−生成(图7F和H)。此外,miR-181a-5p模拟物显著抑制了nHDL诱导的内皮细胞迁移(图8A和E)和血管形成(图8C和G)。miR-181a-5p抑制剂恢复了dHDL抑制的内皮细胞迁移(图8B和F)和血管形成(图8D和H)。这说明nHDL通过抑制miR-181a-5p来激活自噬,从而诱导NO产生和血管生成,而dHDL则通过增加miR-181a-5p并抑制自噬,从而减少NO产生并刺激O2•−生成,以抑制血管生成。
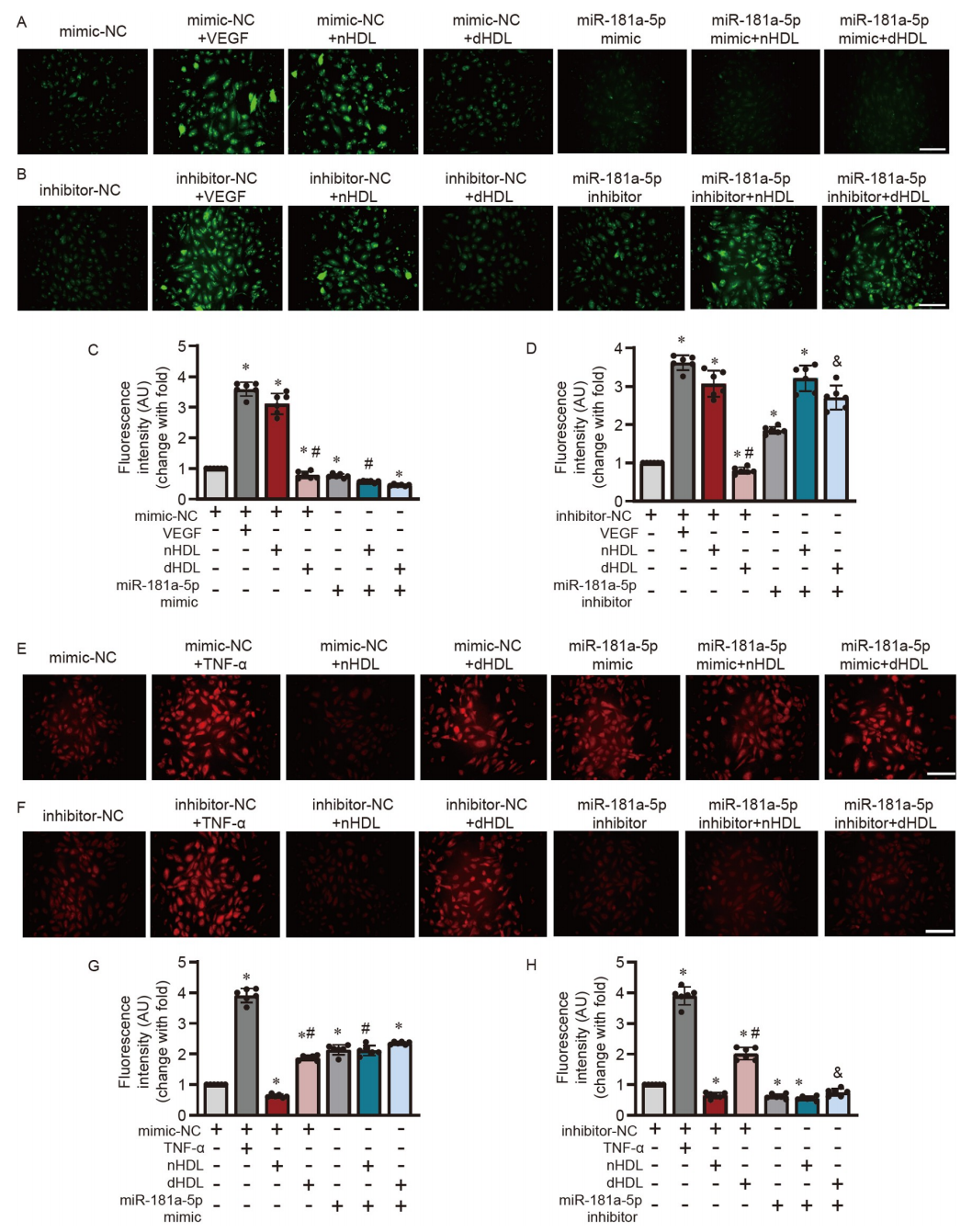
图7. HDL通过调节miR-181a-5p影响HUVECs内皮细胞的NO产生和O2•−生成
A和B,免疫荧光分析显示miR-181a-5p模拟物或抑制剂转染后,HUVECs在nHDL或dHDL刺激下的NO产生;C和D,对A和B中图像的定量分析;E和F,显示miR-181a-5p模拟物或抑制剂转染后,HUVECs在nHDL或dHDL刺激下的O2•−生成;G和H,对E和F中图像的定量分析。
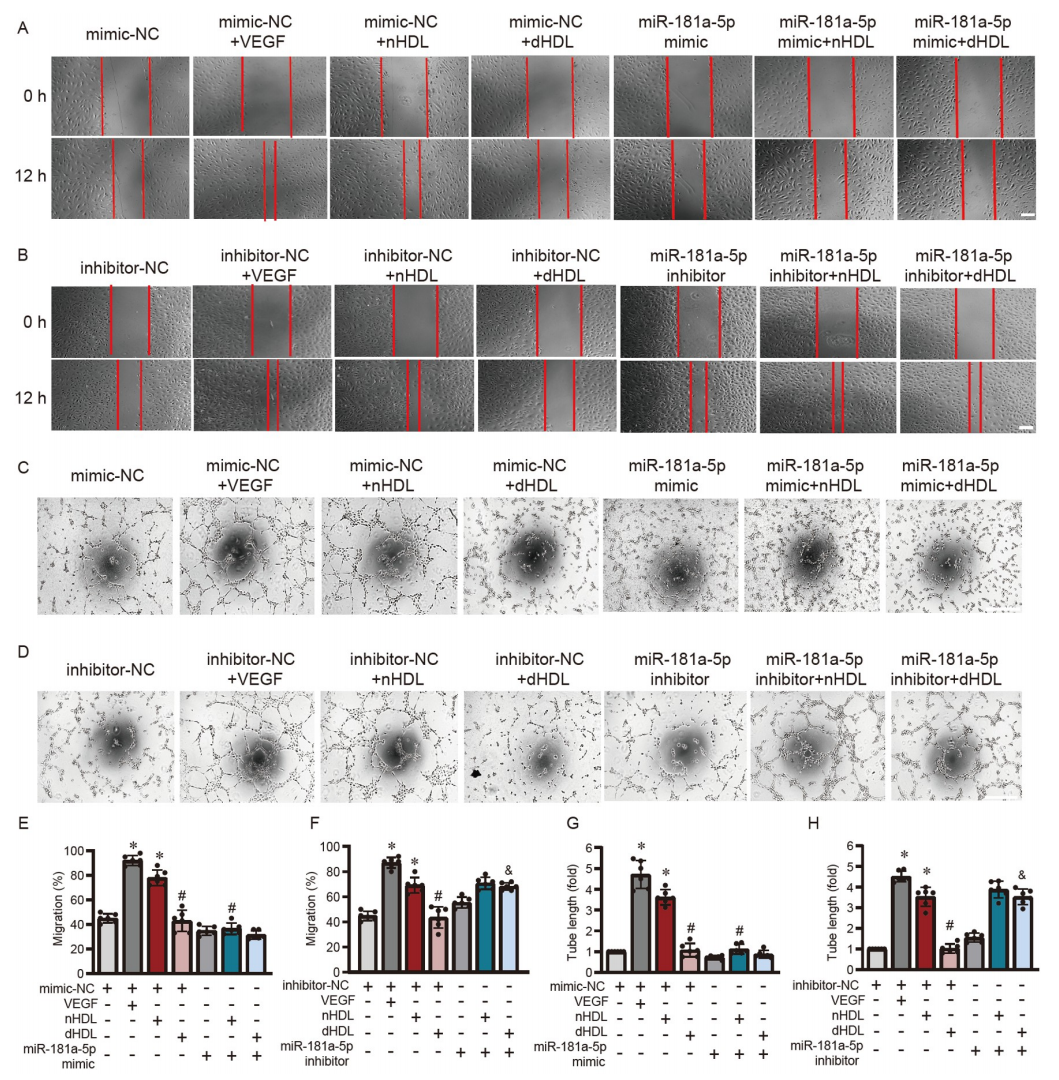
图8. 通过调节miR-181a-5p/ATG5/eNOS轴调控HDL对内皮功能的影响
A和B,显示miR-181a-5p模拟物或抑制剂转染后,HUVECs在nHDL或dHDL刺激下的迁移;C和D,显示miR-181a-5p模拟物或抑制剂转染后,HUVECs在nHDL或dHDL刺激下的血管形成。
ATG5的过表达促进了高胆固醇LDLr−/−小鼠的血管生长
在高胆固醇低密度脂蛋白受体缺失(LDLr−/−)小鼠的HDL中,脂质过氧化物水平明显高于C57BL/6小鼠,表明LDLr−/−小鼠中的HDL具有促炎性。因此,使用高胆固醇LDLr−/−小鼠的缺血下肢来确定自噬对体内血管生长的影响。与C57BL/6小鼠相比,高胆固醇LDLr−/−小鼠的缺血下肢中ATG5和eNOS的表达降低(图9A和B)。过表达ATG5以增加自噬促进了eNOS的表达(图9A和B),改善了缺血下肢的血流(图9C和D)以及侧枝血流(图9E和F)。这些数据提供了在高胆固醇血症中增强自噬可以促进血管生成的体内证据。
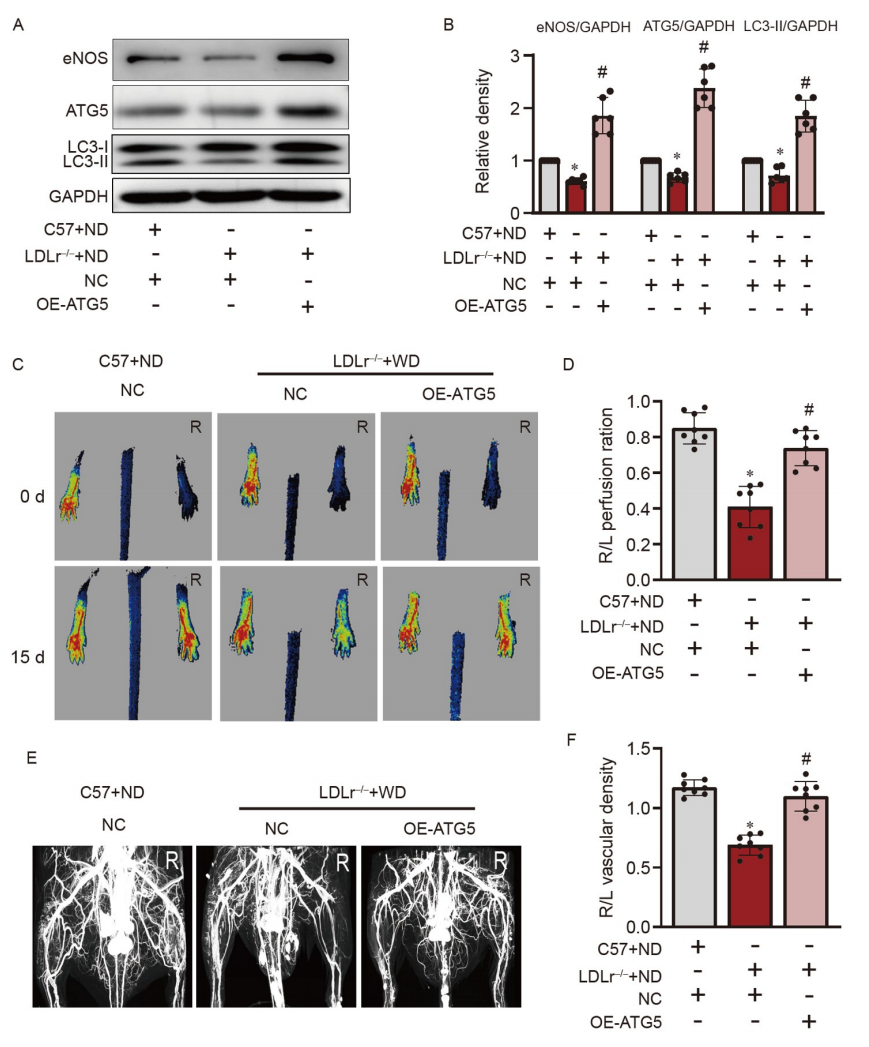
Figure 9. 通过上调ATG5促进体内血管生长
A和B,WB分析显示LDL受体敲除(LDLr−/−)小鼠缺血下肢的胫骨肌中LC3-II、ATG5和eNOS的表达;C和D,血管生成分析的代表性图像和定量分析;E和F,侧枝分析的代表性图像和定量分析。
总结
本研究揭示了nHDL通过调节内皮自噬促进血管生成,而dHDL通过增加miR-181a-5p来抑制内皮自噬,从而减少血管生成。ATG5的过表达能够增加缺血下肢的血流和侧支循环,从而促进血管生成。这项研究首次提供了nHDL通过调节自噬促进血管生成的直接证据,同时也为dHDL通过影响自噬抑制血管生成提供了新的认识。
参考文献:
Bi-Ang Kang, Zhi-Jun Ou, Jing-Song Ou, et al. High-density lipoprotein regulates angiogenesis by affecting autophagy via miRNA-181a-5p. SCIENCE CHINA Life Sciences, 2023.
最新动态
-
04.22
Biology:藏红花花被片中外泌体样纳米颗粒的表征及其免疫刺激活性
-
04.22
酵母建库技术全解析:从原理到实战,助你轻松构建高质量文库
-
04.14
Int J Mol Sci | 植物性外泌体样纳米囊泡治疗早期骨关节炎的分离、表征和体外细胞研究
-
04.14
RNA pull down技术实验的应用场景
-
04.03
RIP技术如何破解RNA蛋白密码?
-
04.03
Food & Function:食用菌纳米囊泡突破-泌阳花菇提取物展现惊人辐射防护力
-
03.27
J. Adv. Res.|植物源性细胞外囊泡治疗IBD:多机制作用与药物载体潜力
-
03.27
植物外泌体:解锁中药未来的 “纳米密码”
-
03.20
细胞内的"GPS"追踪术:揭秘蛋白亚细胞定位的奥秘
-
03.20
BiFC技术,让蛋白互作“看得见”!
 X
X 






