
【客户文章分享】SHMT2 通过 5′UTR 依赖性 ADAM10 翻译启动介导小分子诱导的阿尔茨海默病病理学缓解过程
长期以来,人们一直认为一碳代谢(OCM)与阿尔茨海默病(AD)相关,但潜在的机制仍然不明确。利用化学生物学的优势,作者发现线粒体丝氨酸羟甲基转移酶2(SHMT2)直接调节ADAM金属蛋白酶结构域10(ADAM10)的翻译,后者是AD的治疗靶点。发现小分子Kenpaullone(KEN)通过5'UTR促进ADAM10的翻译,并改善APP/PS1小鼠的认知功能。SHMT2作为KEN的靶基因和5'UTR相互作用的RNA结合蛋白(RBP)介导了KEN诱导的ADAM10在体内外的翻译。SHMT2通过与大量RNA结合控制AD信号通路,并通过与GAGGG基序直接相互作用增强ADAM10的5'UTR活性,而这个基序影响了5'UTR中的核糖体扫描。综上所述,KEN通过将OCM与RNA处理联系起来,展现了治疗AD的潜力,在这一过程中,代谢酶SHMT2通过结合GAGGG基序并促进5'UTR依赖的ADAM10翻译起始,起到了“双重身份”的作用。
研究方法与结果
01、KEN增加了人类和小鼠细胞中的ADAM10表达
KEN已被证实可调节线粒体中parkin的招募,预防听力损失,并增加神经元分化,延长运动神经元的健康寿命。虽然这种分子似乎具有抗癌活性,但在神经退行性疾病中也具有抗凋亡作用。作者首先在SH-SY5Y细胞中测量了ADAM10蛋白水平,发现KEN在0.5至2μM浓度下显著增加了m-ADAM10的表达。进一步研究显示,这种效应不是由于KEN抑制CDKs和GSK-3引起的。而且,KEN还在人类和小鼠细胞中增强了ADAM10蛋白水平,且不引起明显细胞毒性。免疫荧光图像显示,KEN显著增加了SH-SY5Y细胞中ADAM10蛋白的表达水平。此外,KEN还使Aβ42水平显著降低,表明KEN增强的ADAM10在Aβ的产生中具有功能作用。
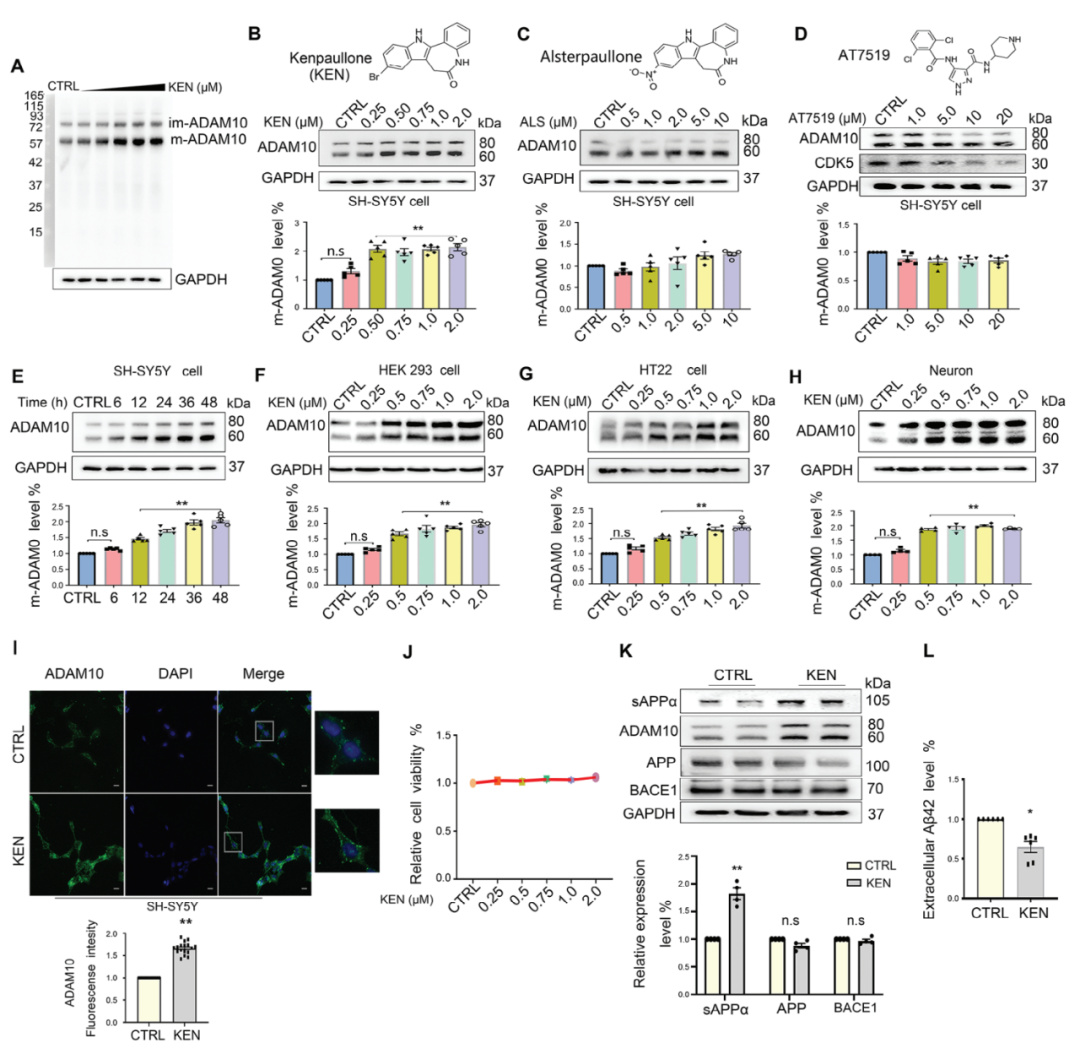
图1: Kenpaullone(KEN)增加了人类和小鼠细胞中的ADAM10蛋白水平
02、KEN介导的ADAM10增强依赖于5'UTR
KEN导致ADAM10增强可能是由于转录、翻译或蛋白质降解的改变,因此作者首先在SH-SY5Y细胞中测量了mRNA水平。如图2A所示,KEN并未改变ADAM10 mRNA水平。转录抑制剂放线菌素D(ActD)或蛋白质合成抑制剂环己亚胺(CHX)单独导致ADAM10蛋白水平降低,而KEN诱导的ADAM10增强在CHX存在时减弱,但在ActD存在时没有减弱(图2B),表明涉及蛋白质合成。看来蛋白质降解机制不涉及这种调节,因为蛋白酶体抑制剂MG132或溶酶体抑制剂氯喹(CQ)未能阻止KEN介导的ADAM10增强(图2C)。此外,作者排除了CDKs/GSK-3在ADAM10调节中的作用,因为单独沉默CDK5或GSK-3β并不改变基础条件下的ADAM10蛋白,并且未能进一步阻止KEN诱导的ADAM10蛋白水平增强(图2D,E)。因此,作者接下来评估了翻译机制是否介导了KEN的效应。如图2F所示,翻译抑制剂4EGI1,它破坏了真核翻译起始因子E(eIF4E)-eIF4G相互作用,显著降低了ADAM10蛋白的基础水平,并进一步减弱了KEN对ADAM10的增强作用。通过转染ADAM10构建,其中包括或删除了5'UTR,作者发现5'UTR的缺失明显增强了ADAM10的基础水平,如先前报道的那样,并且在−5'UTR但不是在+5'UTR中KEN诱导的ADAM10增强被减弱(图2G)。进一步的5'UTR-荧光酶分析显示核苷酸1–144和145–414足以介导KEN的功能(图2H),而BACE1的5'UTR活性未发生改变(图2I),这阐明了对ADAM10的选择性调节。这些结果表明KEN通过5'UTR诱导ADAM10的翻译。
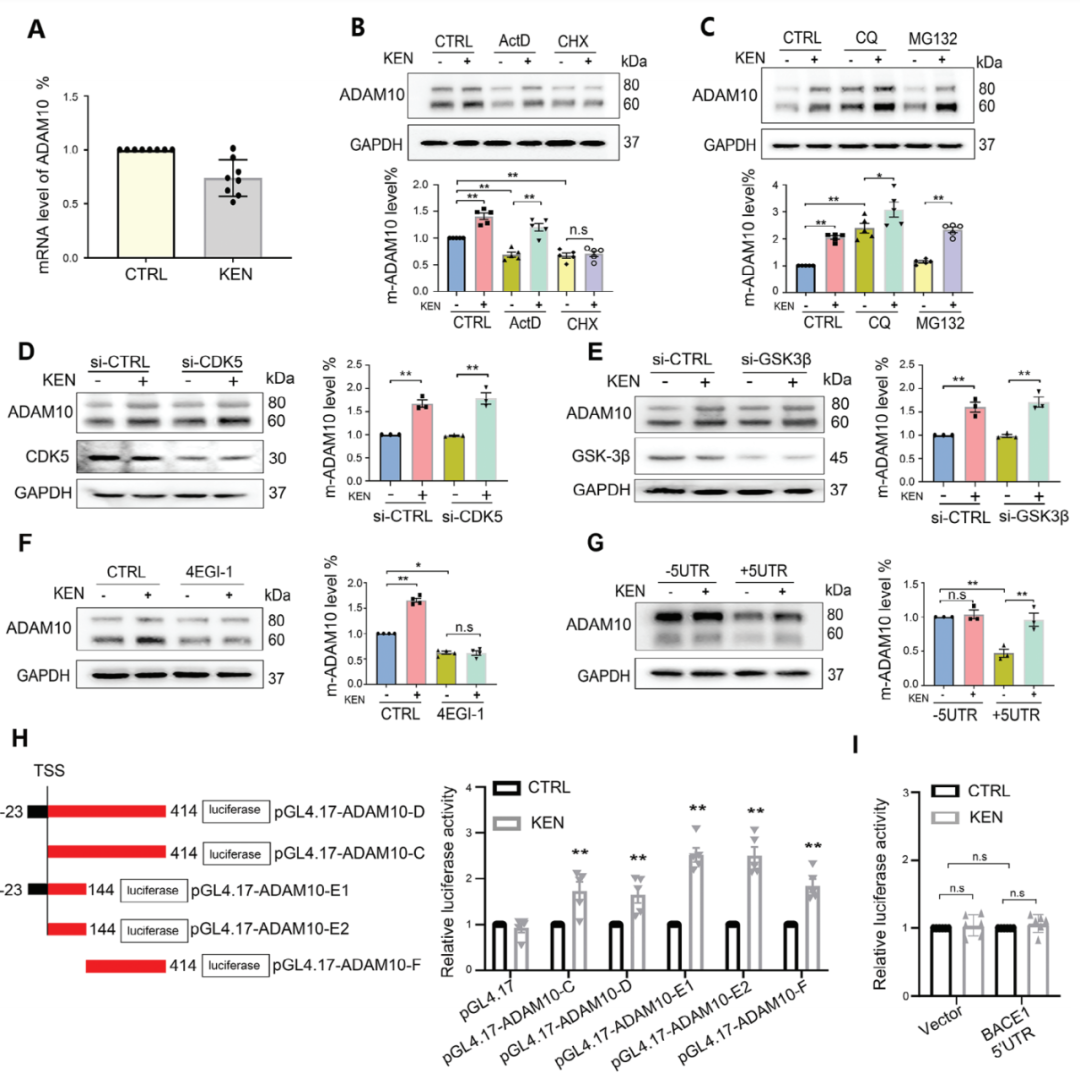
图2. KEN诱导的ADAM10翻译依赖于5'UTR
03、KEN促进了APP/PS1小鼠中ADAM10的表达并改善了认知缺陷
作者接下来确定增强的ADAM10表达是否与体内淀粉样蛋白生成和认知功能的改变相关联(见图3A)。野生型(WT)和APP/PS1小鼠被腹腔注射给予对照组(CRTL)或KEN,从而产生了以下四个组:WT、WT-KEN、APP/PS1和APP/PS1-KEN。鉴于KEN是CDK和GSK-3的抑制剂,这些蛋白激酶已知在选择性位点包括Ser262(Tau262)和Ser396(Tau396)上磷酸化Tau,而这些磷酸化位点在APP/PS1小鼠的大脑中升高。因此,通过测量Tau262/396水平可以验证KEN在大脑中的有效性。此外,ADAM10的底物之一,神经胶质相关细胞粘附分子(NrCAM),用于评估ADAM10激活剂的副作用。如图3B所示,相对于APP/PS1小鼠,APP/PS1-KEN的Tau262/396蛋白水平显著降低,表明KEN成功到达大脑并发挥了生物功能。ADAM10和sAPP的蛋白水平在WT和APP/PS1小鼠中均显著升高,而NrCAM的水平未显著升高,表明KEN选择性地增强了ADAM10而避免了与神经突触生长相关的副作用。作者进一步显示在KEN治疗的APP/PS1小鼠中,ADAM10蛋白水平的增强伴随着Aβ沉积的减少。为了确定KEN是否会影响认知功能,作者使用水迷宫测试和上下文恐惧条件测试评估了APP/PS1小鼠的空间和联想学习记忆。在隐藏平台测试中,APP/PS1-KEN小鼠的逃避潜伏期显著短于APP/PS1小鼠,从第三天开始(见图3E)。在探索试验中,当移除平台时,APP/PS1-KEN小鼠在目标象限停留的时间(见图3F)和穿越目标位置的通过时间(见图3G)显著长于APP/PS1小鼠,表明KEN改善了空间记忆(见图3H)。随后的上下文恐惧条件测试显示,在APP/PS1-KEN小鼠中,冻结的次数(冻结次数)显著增加,冻结的持续时间(冻结时间)显著延长,与APP/PS1小鼠相比(见图3I、J)。WT和WT-KEN之间未观察到显著差异。这些结果表明KEN显著改善了APP/PS1小鼠的空间和联想学习记忆。
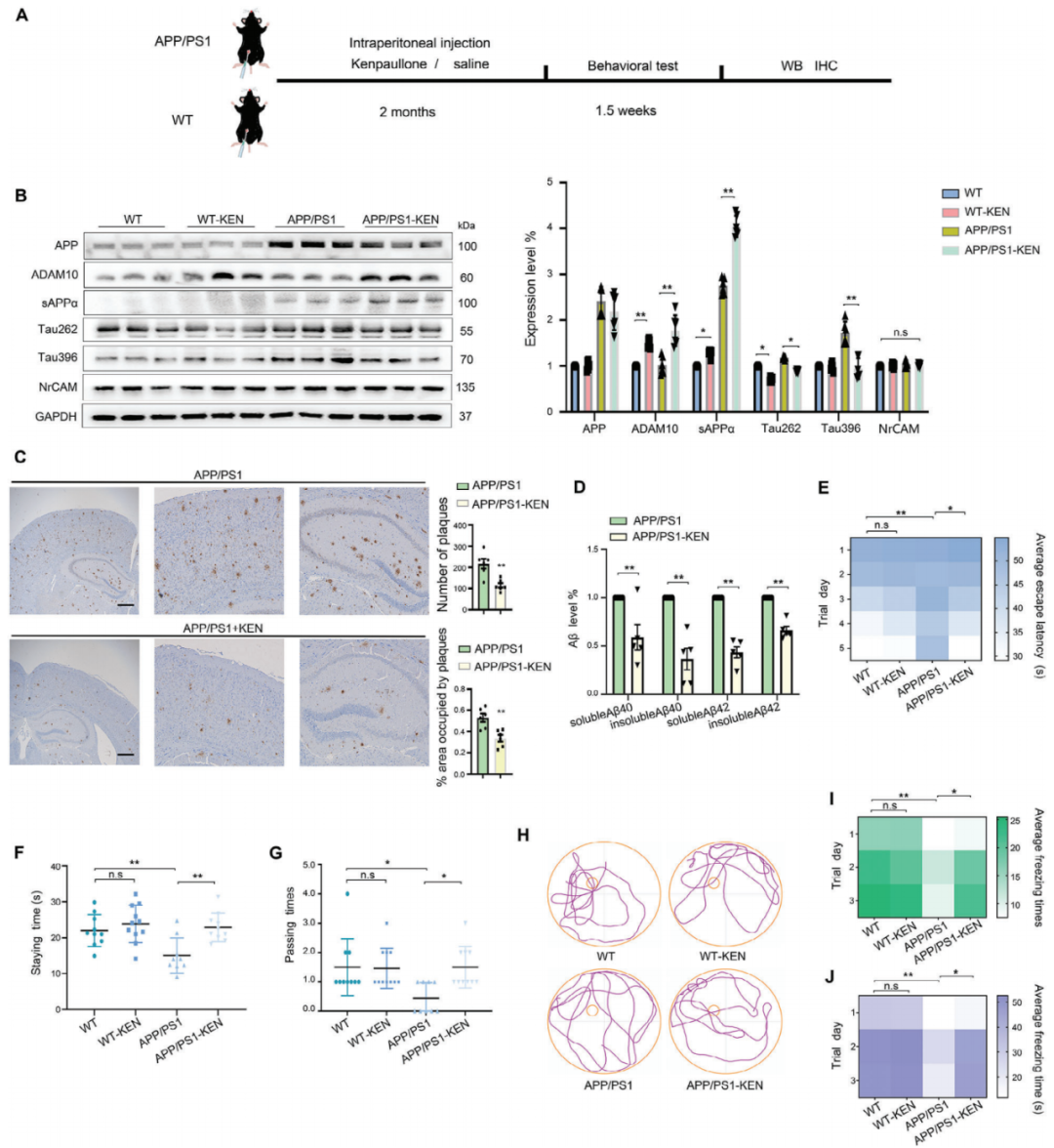
图3. KEN增强了ADAM10并拯救了APP/PS1小鼠的认知缺陷
04、SHMT2是KEN和5'UTR相互作用RNA结合蛋白的靶基因
为进一步理解KEN诱导的ADAM10翻译的潜在机制,作者在SH-SY5Y-APP细胞中通过RNA-seq评估了不同调节的基因(DEGs),其中Aβ过度产生,从而在AD样病理中重塑淀粉样形成。作者构建了共表达网络,以找到在有无KEN的情况下基因之间的关系。KEN诱导了共计733个DEGs,包括296个上调和437个下调的基因。上调的通路包括氨基酰-tRNA生物合成、氨基酸生物合成、碳代谢和叶酸一碳池。重要的是,SHMT2被确定为线粒体OCM中的中心基因。5'UTR的依赖性促使作者推测一些DEGs可能也作为RBPs在调节ADAM10翻译中发挥作用。因此,作者进行了一个RNA结合实验,其中ADAM10的5'UTR被5-溴尿嘧啶标记。作者发现在控制和KEN的情况下有两组RBPs。进一步的蛋白质相互作用分析结果显示,SHMT2与经典的RBPs和核糖体蛋白相互作用。SHMT2与YBX1和CSDE1的相互作用以及5'UTR的进一步验证,说明了SHMT2是靶向5'UTR的RBP网络的关键组成部分。Western blotting实验证实,SHMT2蛋白水平受KEN剂量依赖性显著增加,并且SHMT2敲除阻断了KEN诱导的ADAM10表达增强。进一步的实验表明,干扰SHMT2或5'UTR的小分子破坏了KEN诱导的ADAM10翻译的调节。
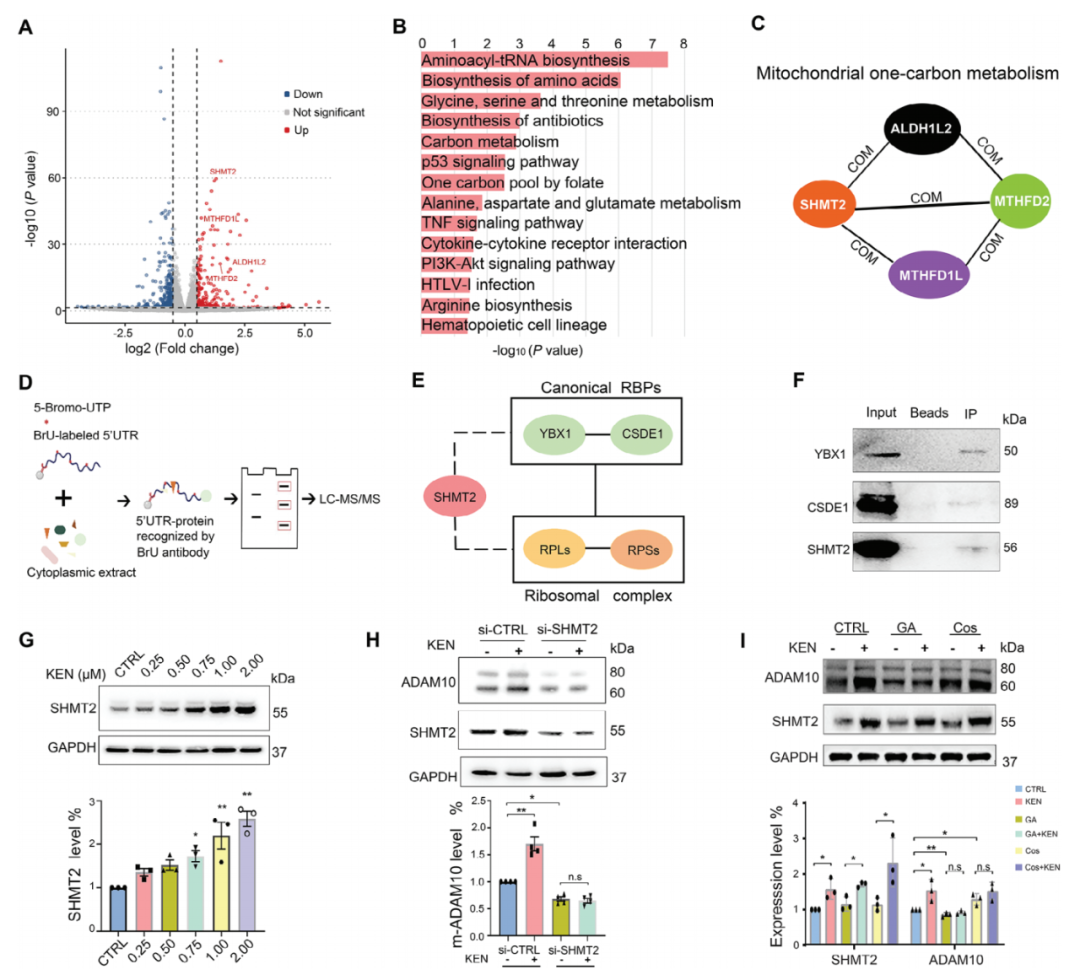
图4. SHMT2 是 KEN 和与 5′UTR 相互作用的 RBP 的靶基因
05、SHMT2敲除减弱了KEN对APP/PS1小鼠中ADAM10和认知功能的影响
作者接着确定了SHMT2是否也介导了KEN对ADAM10和淀粉样物质生成的影响。作者在APP/PS1小鼠的海马区双侧注射病毒AAV载体(对照组)或AAV-shSHMT2,在没有KEN(AAV-shSHMT2-对照组)和有KEN(AAV-shSHMT2-KEN)的情况下,评估了与Aβ负荷和记忆功能相关的ADAM10蛋白水平。如图5A所示,SHMT2蛋白水平的显著降低同时伴随着ADAM10和sAPP水平的显著降低;当SHMT2被沉默时,KEN未能有效增强ADAM10/sAPP。此外,SHMT2的沉默显著增加了海马区Aβ的数量和强度,以及Aβ40/42水平,而这些变化不受KEN的影响(图5B、C)。进一步的行为测试显示,单独SHMT2的沉默显著损害了空间和客观记忆,而额外的KEN未能引起进一步的行为改变(图5D-K)。这些结果表明,在APP/PS1小鼠中,KEN诱导的对ADAM10、淀粉样物质生成和认知功能的调控是通过SHMT2介导的。
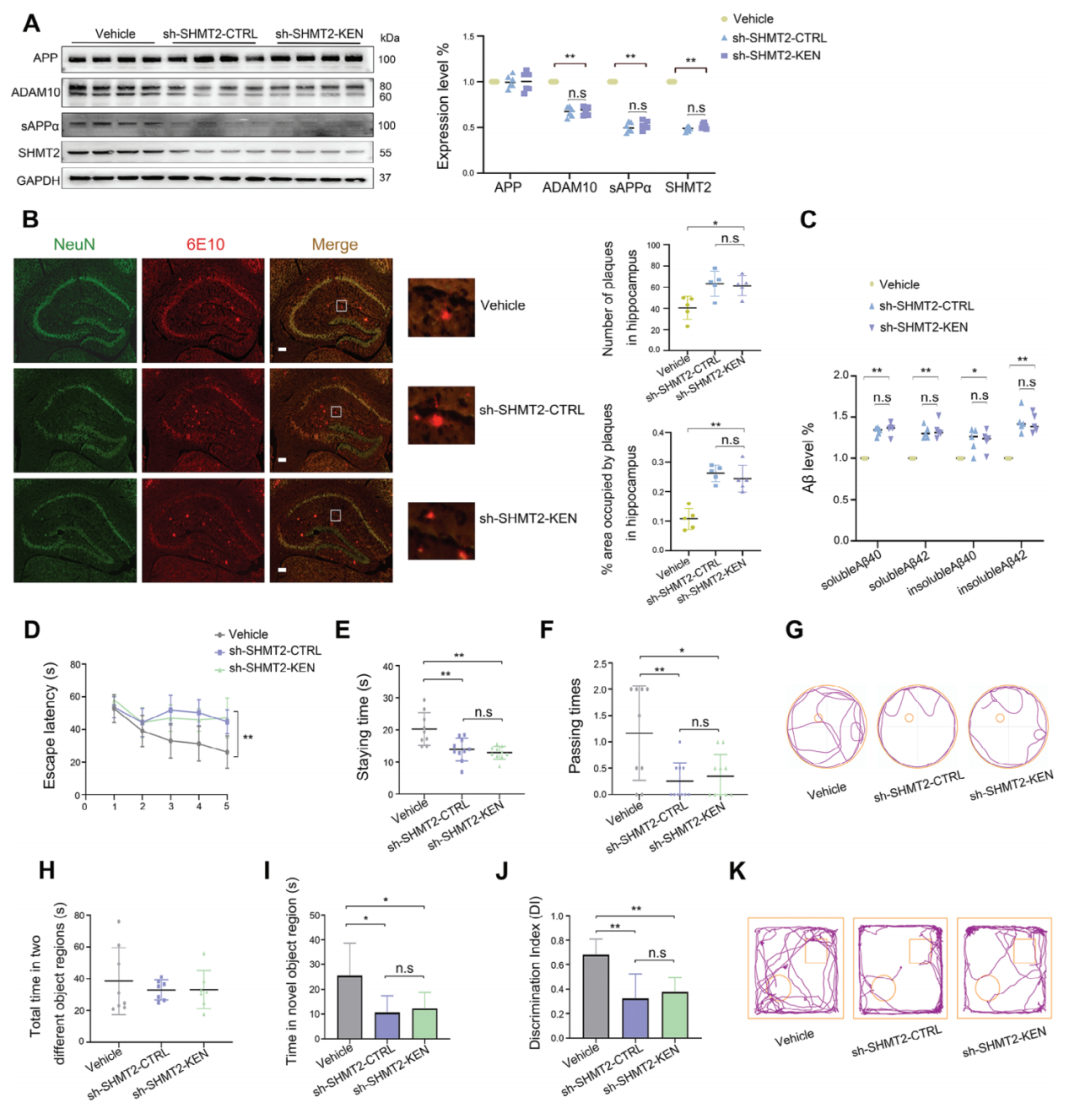
图5. 敲除 SHMT2 可减轻 KEN 对 ADAM10 和 APP/PS1 小鼠认知功能的影响
06、SHMT2结合了大量与AD和KEN的细胞功能密切相关的RNA
RNA结合特性表明SHMT2除了其酶活性外,还可以通过RNA处理来调节细胞功能。因此,作者在SH-SY5Y细胞中使用针对内源性SHMT2的抗体进行了RIP-seq。如图6A所示,SHMT2在对照组和KEN处理组中分别招募了6266和6874个转录本(补充表RIP-Seq)。利用MEME程序进行的5226个在两组中常见的RNA的基序分析显示,SHMT2倾向于与富含GA和GC的基序相互作用(图6A)。重要的是,两组中的KEGG通路均包括核糖体、AD以及多种疾病的神经退行性变化(图6B)。KEN特异性地影响了3401个转录本,包括那些仅在对照组或KEN中独特存在,并且在两组中都被KEN改变;KEGG分析显示了代谢途径和碳代谢与SHMT2功能一致,而其他通路包括RNA转运也被包括在内(图6C)。由于RNA-蛋白相互作用调节了RNA处理,作者推测SHMT2靶向的部分RNA可能会对KEN诱导的细胞功能变化发挥作用。利用KEN的DEGs数据(图4A,B),作者评估了在KEN处理细胞中发生改变的SHMT2靶向mRNA。综合分析显示有112个基因重叠(图6D),GO分析显示转录因子活性和重要的翻译起始因子活性受到影响,表明SHMT2结合的RNA丰度子集特别受到KEN的调节。这些结果表明,SHMT2通过优先与富含GA/GC的序列相互作用,作为RBP发挥作用,这些序列与AD以及KEN的细胞功能密切相关。
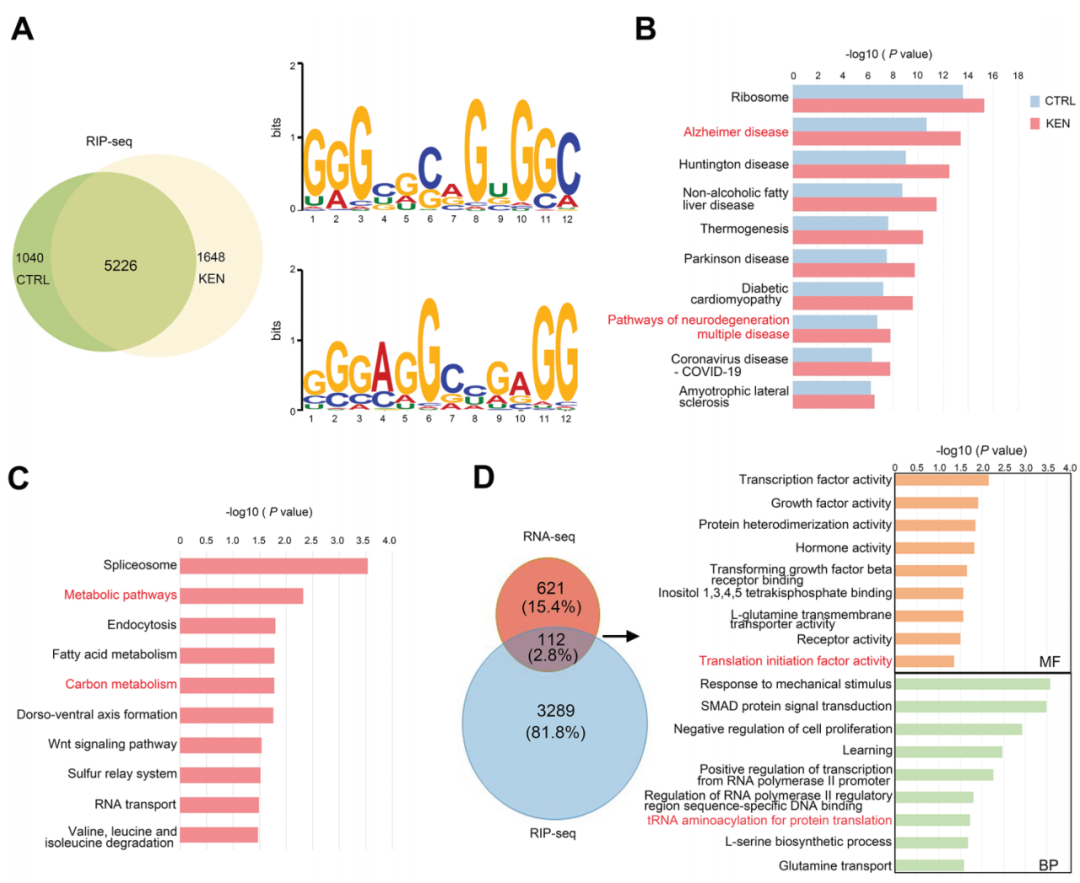
图6. SHMT2靶向的RNA参与了AD和KEN的细胞功能
07、SHMT2通过直接结合GAGGG基序来控制5′UTR活性
为验证SHMT2是否直接结合RNA并发挥其功能,作者使用ADAM10 mRNA的5′UTR不同片段进行电泳迁移实验(EMSA)。如图7A所示,人类全长5′UTR包含多个GAGGG/GAAAG序列,位置已标示。重组的SHMT2蛋白由小泛素样修饰物(SUMO)标记,单独的SUMO不结合任何这些片段(图7B)。SHMT2仅结合包含GAGGG/GAAAG的片段,分别为69–90、156–175、176–200、201–222和223–244。相反,在RNA片段不包括GAGGG/GAAAG基序时,RNA-蛋白复合物(RPC)缺失(图7B)。此外,当对应编号的片段中GAGGG/GAAAG基序发生突变时,未发现RPC(图7C)。作者进一步展示,在100倍过量的非标记RNA存在时,RPC密度显著降低(图7D)。由于KEN还增强了小鼠细胞系和原代神经元中的ADAM10表达(图1),因此作者随后评估了小鼠来源的SHMT2-5′UTR结合。如图7E所示,223nt长的小鼠5′UTR(NM_007399)包含多个GAGGG但不含GAAAG基序。SUMO标记的小鼠SHMT2仅结合包含GAGGG的片段1–29和30–57,而不结合不含GAGGG的片段58–86;而单独的SUMO不结合这些片段。为进一步确认SHMT2是否控制了5′UTR的功能,作者评估了HEK细胞中SHMT2 siRNA的转染后5′UTR活性。如图7F所示,SHMT2敲除显著降低了人类5′UTR的荧光素活性,并显著减弱了KEN诱导的增强效应。这些结果表明,SHMT2直接结合了人类和小鼠来源的GAGGG基序,并调节了5′UTR的活性。
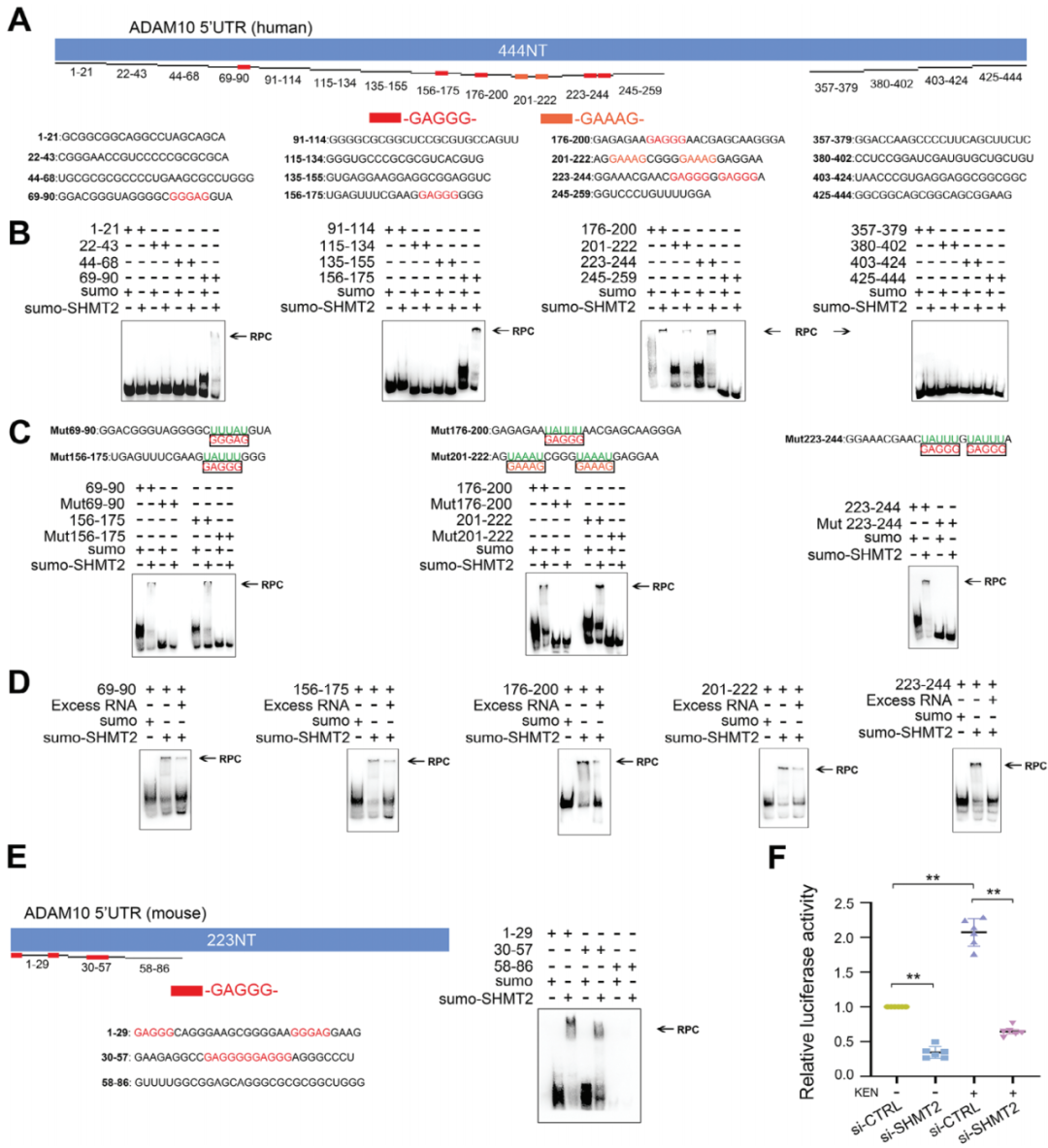
图7. SHMT2 与 GAGGG 基序结合并控制 5′UTR 活性
08、GAGGG突变影响了eIF2的核糖体扫描
SHMT2偏好于GAGGG模体表明cis元素在调控RNA功能中很重要,尤其是ADAM10的5'UTR活性方面。为了弄清楚GAGGG模体在5'UTR功能中的作用,作者将ADAM10 5'UTR中位于26和56核苷酸处的野生型GAGGG突变为GAUUG(mut-ADAM10 5'UTR),并将其克隆到荧光素报告基因构建中。作者预测了相应的二级结构。相比于对照,mut-ADAM10 5'UTR显示出了更高的自由能。令人惊讶的是,GAGGG突变(mut-GAGGG)导致5'UTR的荧光素活性显著增强。作者进一步评估了敲除eIF2S1/2后的5'UTR luciferase活性,结果显示mut-GAGGG的活性仍然显著增加,表明GAGGG模体的突变使得RNA结构发生变化,从而增强了核糖体的扫描。综上所述,GAGGG模体的存在与RNA结构和翻译效率密切相关,SHMT2与GAGGG模体的结合可能改变RNA结构,促进ADAM10的翻译,从而减少淀粉样蛋白生成。
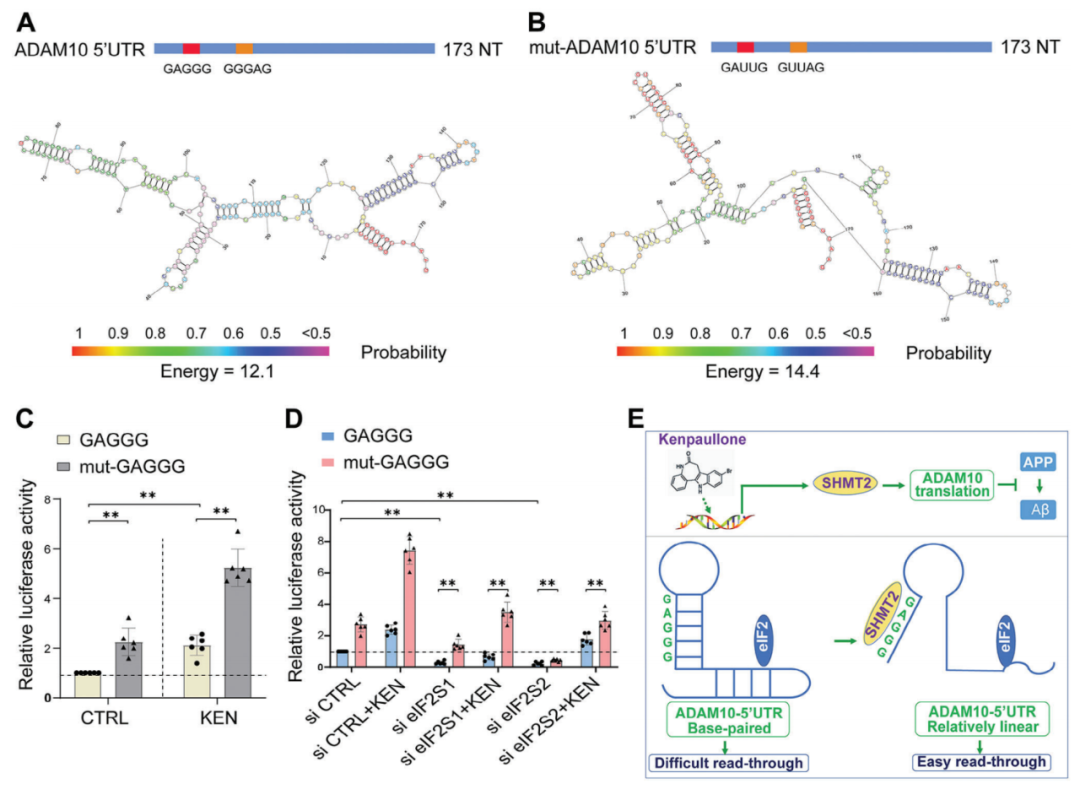
图8. GAGGG 突变缓解了 eIF2 敲除对 5′UTR 活性的抑制
结论
本研究揭示了SHMT2在调节与AD相关的RNA修饰中的作用,尤其是在KEN对ADAM10 5'UTR活性的影响中的介导作用。然而,SHMT2在调节多种生物学途径方面的多面作用不支持将KEN-SHMT2的关联作为治疗AD的可能途径,减少淀粉样生成只是SHMT2多种效应之一。此外,KEN在动物模型中改善的认知功能也涉及到对Tau病理的减少,这可能与SHMT2无关,因为KEN是已知的GSK和CDK的抑制剂,这些激酶被认为是磷酸化Tau的。SHMT2在AD病理生理学中的详细功能仍有待进一步澄清。
在本文中,重组SHMT2蛋白由金开瑞生物提供。

最新动态
-
04.20
分子对接从入门到进阶:“这5张图你必须看懂”!
-
04.20
siRNA实验从入门到精通:合成与转染的那些“坑”你踩过吗?
-
04.20
酵母文库构建技术解析:如何高效筛选功能基因?
-
04.20
【客户文献分享IF:33】重庆医科大学赵金秋团队揭示肝内胆管癌免疫逃逸新机制:YAP通过转录抑制RNF125重塑肿瘤免疫微环境
-
04.20
国自然倒计时:蛋白互作研究还在只做Co-IP?这8种技术让评审专家“无刺可挑”
-
04.20
研究合集:中草药囊泡-生姜/白术/黄芪/半夏/鱼腥草/无花果/铁皮石斛等-36个经典案例!
-
04.07
【客户文献分享】药用真菌槐耳(Huaier)来源外泌体样纳米颗粒:携带miRNA跨物种调控,精准抑制乳腺癌
-
03.24
IF:12.6首报!黄芪来源外泌体样纳米颗粒三重机制协同治疗前列腺癌
-
03.11
同为抗体,为什么一抗看“抗原种类”,二抗却看“一抗种属”?
-
03.11
J Nanobiotechnology(IF=12.6):蔓越莓“纳米邮差”递送麦角甾醇,重启早衰卵巢功能!
 X
X 






