
酵母双杂交建库-试剂盒操作流程解析及常见问题解答
酵母双杂交建库实验是研究蛋白质相互作用中的一个很重要的实验,首先我们要根据蛋白质的定位,选择不同的建库方式。如果蛋白质定位在细胞核上,是基于GAL4酵母单双杂的系统,使用载体PGADT7进行酵母建库;如果蛋白质是定位在细胞膜上,则是基于分离的泛素(split-ubiquitin)介导的膜蛋白酵母双杂交系统,使用pPR3-N载体进行酵母文库的构建。所以大家一定要学会“对症下药”,才能保证实验顺利进行。
既然选好了方向,那我们就要开始正式建库啦。有的小伙伴会经常遇到建库失败的问题,别担心,小金不仅给大家提供了超级好用的酵母建库试剂盒,还给你们总结了实验过程中需要注意的关键细节,大家赶紧进来抄作业吧!
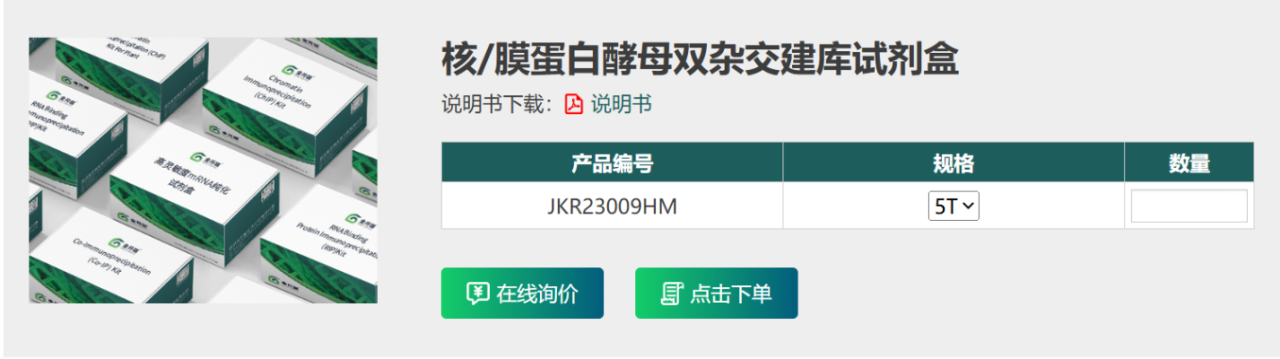
一、酵母建库实验流程图
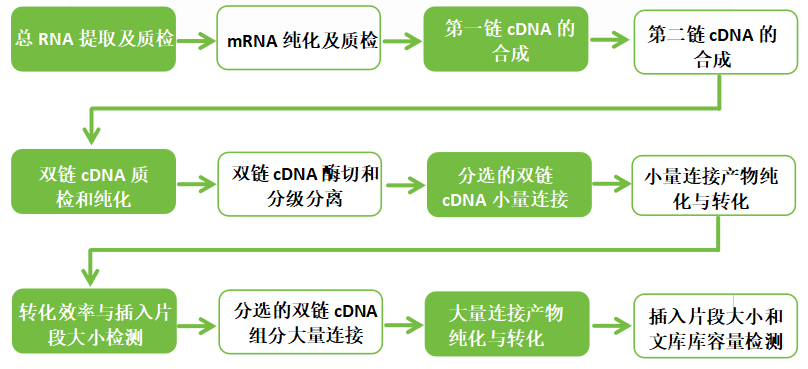
二、酵母建库实验关键点
1、RNA提取与质量检测(源头把控)
总RNA起始材料的完整性和纯度是高质量 cDNA 合成的重要元素,一份高纯度的RNA是实验成功的关键,我们可以根据自己的实验材料选择合适的方法提取RNA,确保RNA电泳图中无降解或污染。RNA电泳图如下:
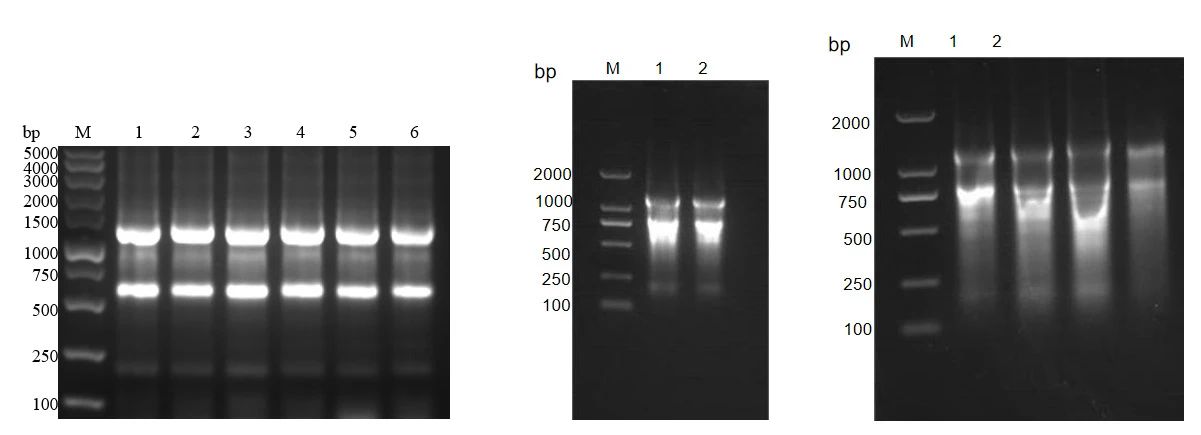
总RNA跑胶示例图
左图总RNA样品是合格的,中间和右图总RNA样品不合格
2、mRNA纯化及质检
建议使⽤经过纯化的mRNA作为起始材料来进⾏单双链合成,因为纯化的mRNA可以排除rRNA和tRNA对下游PCR扩增产⽣的⾮特异性⼲扰,增强⽂库的有效扩增。使用mRNA purification kit将mRNA从100μg~500μg完整度良好的总RNA中进⾏分离纯化。
3、第⼀链cDNA的合成
在配备溶液体系的时候,需要保证RNA的用量,计算方法:X=总RNA加1μg,浓度至少为0.1μg/μL。mRNA至少加0.3μg以上,浓度至少为0.1μg/μL),在合成单链cDNA后,需保存在-20℃,建议三个月内使用,过期需要重新合成。
|
总RNA/mRNA样品 |
XμL |
|
CDSⅢ引物(10μM) |
1μL |
|
dNTP Mix(10mM) |
1μL |
|
无菌H2O |
11-X μL |
|
总体积 |
13μL |
4、第二链 cDNA 的合成
这步实验中,可以先扩增50 μL体系,合成的双链cDNA进行琼脂糖凝胶电泳检测,确认合格后,再将剩下的一链cDNA全部合成二链cDNA 。
5、双链cDNA质检和纯化
使用PCR-Pure Kit纯化合成的双链cDNA,纯化前一定要检查PW buffer试剂中是否加入无水乙醇,若忘记加入,有可能会导致实验失败。双链的合成实验尽量要在一天之内完成,放置时间太久,可能会影响实验效果。
6、双链cDNA酶切和分级分离
酶切过程中要注意,将配备好的酶切体系用移液器轻轻抽打混匀,并且短暂离心确保溶液集中在管底。双链cDNA只需要用Sfi I酶进行单酶切,通常是1μL切2μgDNA,做一次建库需要切10μg的纯化后的双链cDNA,有剩下的可以先保存。
分离过程中将离心柱反转几次,去除离心柱中的气泡,同时要控制缓冲液通过离心柱中的流速,如果流速太慢或一滴的体积太小,则需重新重悬基质并重复滴注程序直至达到正常参数。
7、分选的双链 cDNA小量连接
连接体系如下,注意cDNA和线性化载体按照3:1的比例添加,连接时间大约是16小时左右(时间不宜过短)。
|
双链cDNA |
两者摩尔比建议3:1 |
|
PGADT7-SG1,2,3线性化载体(核体系)或pPR3-NS 1,2,3线性化载体(膜体系) |
|
|
10 ×T4 DNA Ligase Buffer |
1μL |
|
T4 DNA Ligase |
1μL |
|
去离子水 |
补足至10μL |
8、小量连接产物纯化与转化
小量连接转化时,连接产物3μL+97μL DH10B感受态,且小量连接转化只需要鉴定片段长度,所以可以直接涂含有Amp抗生素的小平板,每个涂50-80μL左右。
9、转化效率和插入片段大小检测
我们可以从每个组份扩10个菌,跑一下琼脂糖凝胶电泳,判断一下片段长度是否符合要求,确认合格后方可进行下一步实验。
10、分选的双链cDNA组分大量连接
大量连接一定要用新鲜的DH10B感受态。而且涂板时尽量不要离心,正着培养,避免大肠菌最后扒在板子上刮不下来,1mL 涂1个大平板(玻璃平板倒厚一些会比较容易收库,塑料平板比较薄很容易收不下来)。
11、大量连接产物纯化与转化
连接产物涂板37℃温箱过夜后,需要根据每个平板的克隆数,铺板体积以及稀释因子来计算文库滴度。在获得原始文库菌液时,需要加入等体积的灭菌的50%甘油,保存于﹣80℃冰箱。用如下方法统计文库滴度,克隆数 = cfu/mL 铺板体积 (mL) ×稀释因子(注意 : 由于移液器的误差,2~5 倍的滴度差异是正常的)。
• 每板克隆数 = 100
• 铺板体积 = 0.1 mL
• 稀释因子 = 10-4
则文库滴度为:
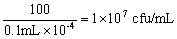
12、插入片段大小和文库容量检测
挑单克隆进行菌落 PCR,抽样检测文库基因插入片段大小,大量文库鉴定一般扩20个左右,可以选10个送去测序,核对种属是否正确。
三、酵母文库检测结果展示
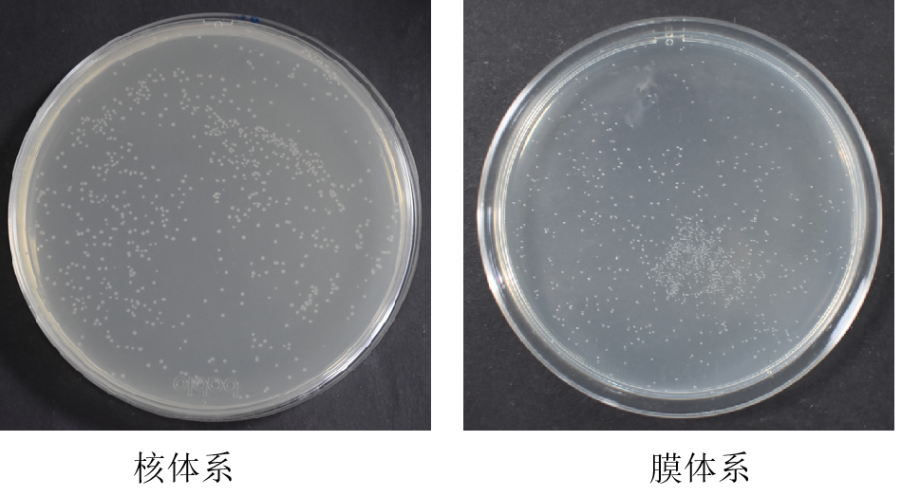
1、试剂盒构建的酵母文库库容量检测结果
2、菌落PCR鉴定结果
核酵母杂交文库构建:文库插入片段PCR鉴定70%在750bp以上,文库的阳性率>90%
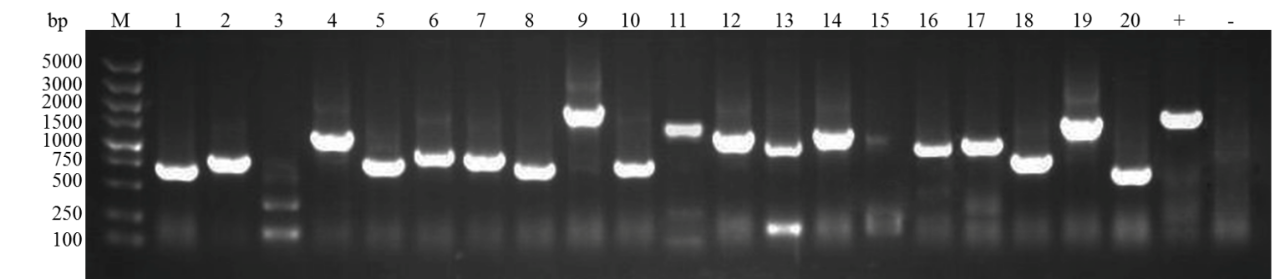
核体系:+:以PGADT7-T为模板
-:以水为模板
膜酵母杂交文库构建:文库插入片段PCR鉴定80%在750bp以上,文库的阳性率>90%
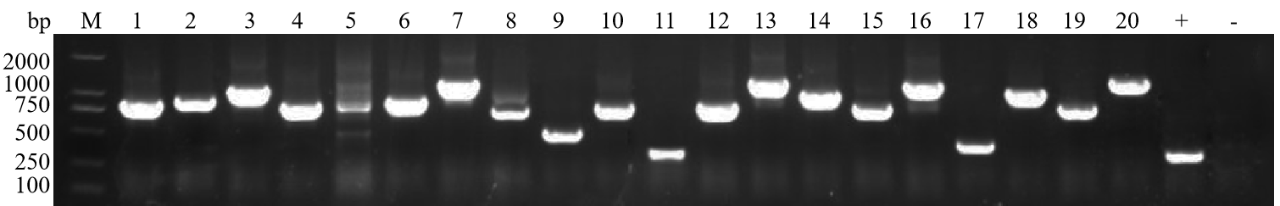
膜体系:+:以pPR3-N为模板
-:以水为模板
四、常见问题与解答
Q1、双链cDNA跑胶条带小于预期大小
使用的总RNA或者mRNA存在降解、样品不纯、浓度太低等问题。可以通过以下方法解决:
1、通过琼脂糖凝胶检测总RNA和mRNA的质量,确保质量和纯度均没有问题(如果RNA质量很好,但浓度太低,则使用更多的RNA来进行双链的合成;如果RNA质量不行,则需要重新提取RNA);
2、对RNA的稳定性进行检测,如果RNA容易降解,检查所用的离⼼管、tip头等是否有RNase的污染,并换⼀种方法重新提取总RNA;
3、为了更容易地确定是否是RNA的问题,请使用对照RNA进行平行反应。
Q2、双链cDNA产物中存在较多<0.1Kb的条带?
可能是PCR的循环数太多了,重新使⽤第⼀链产物进⾏第⼆链的合成PCR时减少3~4个循环数。
Q3、如何检测文库质量,文库容量低应该怎么解决?
对于我们提供的大肠杆菌文库甘油菌,可以进行梯度稀释后铺板培养,第二天计算库容量,同时挑取单克隆进行PCR扩增,以判断插入片段长度和空载率。如果检测出文库容量低,有可能是连接效率低,可以通过以下两种方式解决问题:
1、检查分级分离后双链cDNA的浓度,双链cDNA的浓度应该在100~200 ng/μL的浓度范围内。
2、按照试剂盒说明书中的连接体系,连接体系中线性化载体和双链产物的配⽐不合适,或者连接体系中这两者所加⼊的量太多均会影响连接效率。
Q4、DNA浓度不足,提取的DNA浓度太低,无法满足文库构建的要求。
可以尝试使用其他DNA提取方法来提高DNA浓度,如酚/氯仿法或商业DNA提取试剂盒。此外,可以使用PCR进行DNA预增强,以增加可用DNA的量。
Q5、⽂库插⼊⽚段太⼩?
如果超过50%的克隆具有较小的插入⽚段(即<0.4kb),则双链cDNA分级分离不成功。
1、重新合成双链cDNA、纯化后酶切双链cDNA进行分级分离;
2、严格执行分级分离的实验步骤,例如,在洗涤和洗脱步骤中使用过多或过少的柱缓冲液,或省略⼀个步骤,则柱的分级分离功能将减弱;
3、不要让柱中的基质在洗涤等步骤中变干,干燥的基体可以从柱壳体的内壁收缩
离开。然后,双链cDNA混合物可以沿着柱的侧面向下流动,使小污染物过快地进入基质主体,并在早期的级分中洗脱;
4、柱应在室温下储存和使用。如果在4℃下冷却,然后加热到室温使用,可能会形成气泡,从而干扰柱的正常工作;
5、双链cDNA混合物在柱表面上的极端不均匀沉积会导致双链cDNA与低分子量污染物的低效分离。
最新动态
-
04.30
Journal of Nanobiotechnology:苦瓜来源的外泌体样稳定p62表达以改善阿霉素心脏毒性
-
04.30
Theranostics-源自水果、蔬菜和草药的类外泌体纳米颗粒:治疗和药物递送的创新策略
-
04.22
Biology:藏红花花被片中外泌体样纳米颗粒的表征及其免疫刺激活性
-
04.22
酵母建库技术全解析:从原理到实战,助你轻松构建高质量文库
-
04.14
Int J Mol Sci | 植物性外泌体样纳米囊泡治疗早期骨关节炎的分离、表征和体外细胞研究
-
04.14
RNA pull down技术实验的应用场景
-
04.03
RIP技术如何破解RNA蛋白密码?
-
04.03
Food & Function:食用菌纳米囊泡突破-泌阳花菇提取物展现惊人辐射防护力
-
03.27
J. Adv. Res.|植物源性细胞外囊泡治疗IBD:多机制作用与药物载体潜力
-
03.27
植物外泌体:解锁中药未来的 “纳米密码”
 X
X 






