
【干货】手把手教你做酵母双杂交点对点验证
酵母双杂交及系统是一种鉴定和检测蛋白质相互作用的研究方法,因蛋白的定位不同又分为核蛋白酵母双杂交和膜蛋白酵母双杂交。有小伙伴会问,两者在实验过程中到底有什么区别?点对点实验到底怎么做?为了给大家答疑解惑,小金进行了详细的总结!
核体系酵母双杂交:基于GAL4系统,主要运⽤该系统实现筛选得到调控⽬的基因启动⼦的转录因⼦、或者筛选和检测细胞核、细胞质蛋⽩之间的蛋⽩—蛋⽩相互作⽤。(无需选择载体)
膜体系酵母双杂交:是基于分离的泛素(split-ubiquitin)介导的膜蛋⽩酵⺟双杂交系统,该系统可用于膜蛋⽩-膜蛋⽩或膜蛋⽩-胞质蛋⽩之间的互作检测。
膜蛋白双杂交点对点实验开展前期最基本的要求就是Bait蛋白的表达必须在膜上准确定位,而且Cub-LexA-VP16和Prey(NubG)必须位于胞浆中,整个系统才能正常运行。因此需要对诱饵蛋白精确定位后选择合适的载体。
1.那我们如何预测蛋白的定位呢?
小金来给大家推荐几个网站:
CDS序列翻译为氨基酸序列:https://web.expasy.org/translate/
蛋白跨膜结构域预测:https://www.detaibio.com/tools/transmembrane.html
蛋白信号肽预测:https://urgi.versailles.inra.fr/predotar/
蛋白结构域预测:http://smart.embl.de/
2.预测了蛋白的定位后载体该选择呢?
请看下方!
膜体系酵母双杂交诱饵载体选择
|
诱饵N端 |
诱饵C端 | 诱饵载体 |
|
胞外且含有信号肽 |
胞内 | pBT3-SUC |
|
胞内 |
胞外 | pBT3-N |
|
胞内 |
胞内 | |
|
胞外且不含有信号肽 |
胞内 | pBT3-STE |
|
胞内 |
胞内 |
注:若N端和C端都位于胞外,可截短使得C端位于胞内段,再选择相应的载体。
一、实验流程
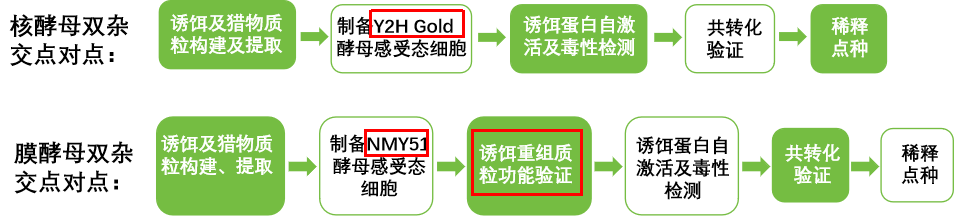
两者区别
➢感受态细胞不同,核酵母使用Y2H Gold,膜酵母采用NMY51。
➢膜酵母双杂交点对点实验增加了一个诱饵重组质粒功能验证。
为什么膜酵母体系需要增加诱饵重组质粒功能验证呢?
原因是:膜系统载体的选择主要根据诱饵跨膜的类型进行选择,为验证诱饵载体是否构建正确,能在细胞中形成正常的功能,需要将重组诱饵质粒和猎物载体pOST1-NubI共转化酵母菌株中,通过涂布缺陷型和报告基因平板,如果均生长,说明诱饵和猎物的功能域形成了完整的泛素,激活了报告基因的表达,诱饵构建无误,能够形成正常的定位和功能。
二、实验步骤
以膜酵母双杂交为例详细介绍点对点实验方法:
1.诱饵质粒、猎物质粒构建及提取
(1)将构建好的诱饵质粒pBT3- bait转化至大肠杆菌株中,涂布于LB平板(卡那抗性)上,37 ℃培养过夜;
(2)将构建好的猎物质粒 pPR3- prey转化至大肠杆菌株中,涂布于LB平板(氨苄抗性)上,37 ℃培养过夜;
(3)挑选含重组质粒的单菌落至3 mL LB液体培养基中,37 ℃培养过夜;
(4)取700 μL菌液保种,剩余菌液接种于300 mL LB液体培养基中,37 ℃培养过夜;
(5)收集菌体并进行质粒大量抽提。
注:抽提质粒浓度需达到200ng/μL。
2.制备NMY51酵母感受态细胞:
(1)将酵母NMY51划线于YPDA固体培养基,30 ℃倒置培养3 d;
(2)挑单菌落于10 mL YPDA液体培养基中,30 ℃,220 rpm培养16 h;
(3)取1 mL菌液于100 mL YPDA中,30 ℃,220 rpm培养8~12h至OD600值为0.4~0.6;
(4)700×g离心5 min,弃上清;
(5)以30 mL预冷去离子水重悬菌体;
(6)700×g离心5 min,弃上清,以1.5 mL预冷的 TE/LiAc重悬菌体;
(7)高速离心15 s,弃上清,以600 μL预冷的 TE/LiAc重悬菌体,分装后置于冰上备用。
3.诱饵重组质粒功能验证:
(1)将carrier DNA置于沸水中煮 5min,立即置于冰上2min,如此重复一次;
(2)按照以下组份,分别配制转化体系:
|
诱饵重组质粒功能验证 |
|
4μg pBT3-Bait诱饵质粒 |
|
4μg pOst1-NubI质粒 |
|
100μL NMY51酵母感受态细胞 |
|
10μL carrier DNA |
缓慢轻柔加入500 μL PEG/LiAc,混匀颠倒;
(3)30℃水浴30min,每10min轻柔混匀一次;
(4)加入20 μL DMSO,轻柔混匀;
(5)42℃水浴15min,每5min轻柔混匀一次;
(6)800g离心1min,弃上清,加入800 μL YPDA重悬,封口膜封上;
(7)30℃,150rpm,振荡培养1.5h;
(8)800g离心5min,弃上清,加入1 mL 0.9% NaCl重悬,分别涂150ul于DDO、TDO、QDO平板;
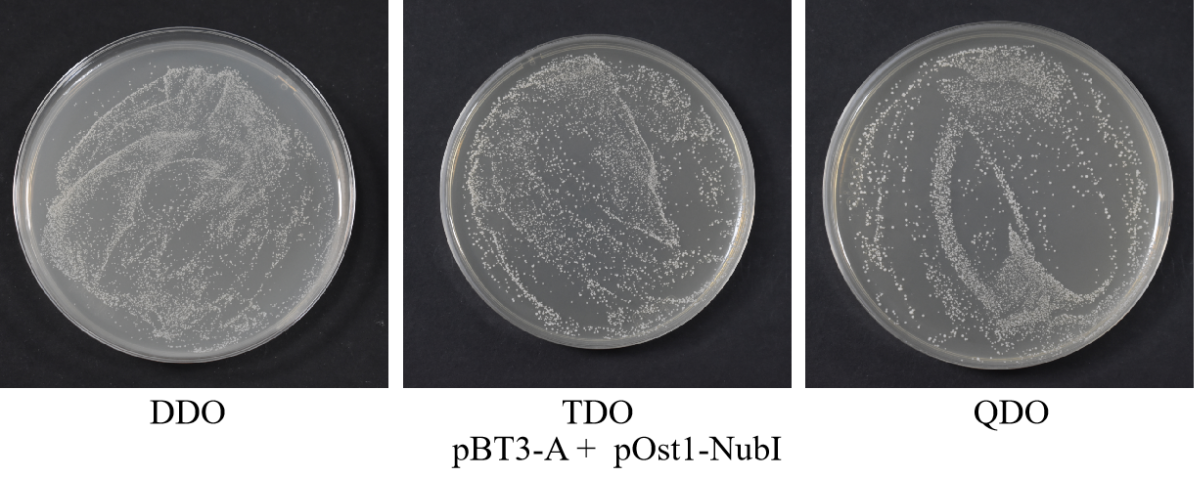
(9)30 ℃倒置培养3-5 d,观察菌落数量及状态。DDO上有菌落生长,说明重组诱饵质粒成功转入宿主菌且对宿主无毒性。在DDO上菌落生长正常的情况下,观察TDO和QDO上菌落生长状况,平板 TDO 和 QDO 均有菌落生长,说明诱饵质粒能够适应 ubiquitin系统,功能验证结果为阳性,可以进行下一步实验。如果平板TDO和QDO上无菌落生长,则诱饵载体构建有问题,请重新检查蛋白定位情况,选择合适的载体进行诱饵载体构建。
注:Nub I为野生型的ubiquitin的N端结构,保留了原始的三个I 氨基酸结构,可以在不需要两个蛋白产生相互作用的情况下两个C 端和N端的ubiquitin结构域结合在⼀起产生具有功能的ubiquitin,从而激活下游的报告基因。如果将pBT3-Bait和pOST1-NubI 质粒共转化NMY51,功能验证结果为阳性,可以排除由于诱饵蛋白造成的空间位阻而产生假阴性,同时能说明Bait(Cub-LexA-VP16)跨膜蛋白正常表达,因此能够确认诱饵重组质粒可以应用于下游的点对点验证。
注:良好的菌落生长状态一般是圆形凸起、边缘整齐且光滑湿润、不透明,为乳白色偏浅黄的菌落,其次有酵母气味。
诱饵A蛋白毒性检测和功能验证结果
4.诱饵蛋白自激活及毒性检测
(1)按上述步骤制备NMY51感受态细胞;
(2)将carrier DNA置于沸水中煮 5min,立即置于冰上2min,如此重复一次;
(3)按照以下组份,分别配制转化体系:
|
自激活及毒性检测 |
|
4μg pBT3- bait诱饵质粒 |
|
4μg pPR3-N质粒 |
|
100μL NMY51酵母感受态细胞 |
|
10μL carrier DNA |
(4)缓慢轻柔加入500 μL PEG/LiAc,混匀颠倒;
(5)30℃水浴30min,每10min轻柔混匀一次;
(6)加入20 μL DMSO,轻柔混匀;
(7)42℃水浴15min,每5min轻柔混匀一次;
(8)800g离心1min,弃上清,加入800 μL YPDA重悬,封口膜封上;
(9)30℃,150rpm,振荡培养1.5h;
(10)800g离心5min,弃上清,加入1 mL 0.9% NaCl重悬,分别涂150ul于DDO、TDO、TDO/ 3-AT 5 mM、TDO/ 3-AT 10 mM、TDO/ 3-AT 15 mM、QDO平板;
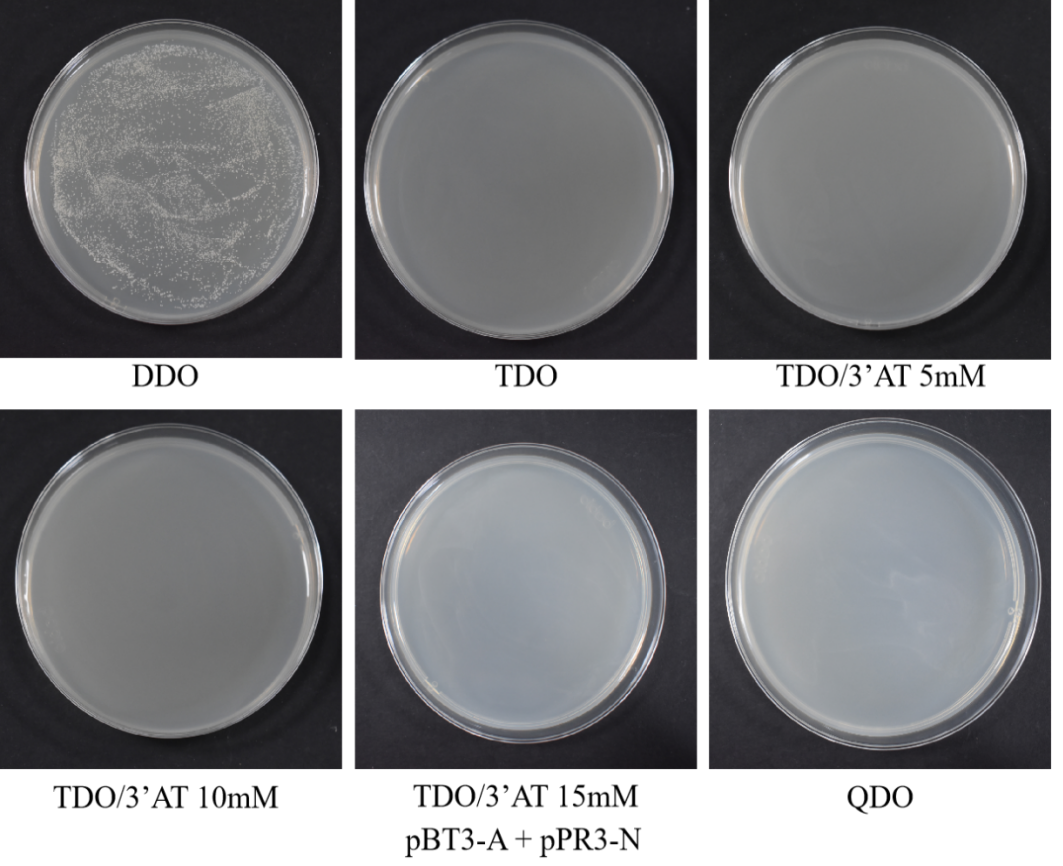
(11)30 ℃倒置培养3-5 d,观察菌落数量及状态,DDO上有菌落生长,说明重组诱饵质粒成功转入宿主菌且对宿主无毒性。在DDO上菌落生长正常的情况下,观察TDO、TDO/ 3-AT 5 mM、TDO/ 3-AT 10 mM、TDO/ 3-AT 15 mM上菌落生长状况, 3-AT浓度越高,抑制自激活的能力越强,平板开始不长菌落则说明在该浓度下诱饵质粒不能自激活酵母细胞报告基因,后续共转验证可用该浓度的3-AT。
诱饵A蛋白自激活检测结果图
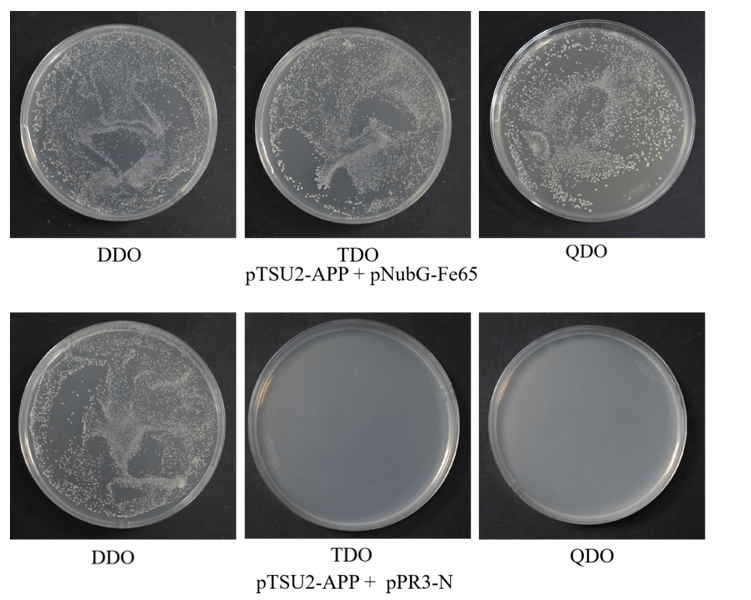
5.共转化验证
(1)准备NMY51感受态细胞;
(2)将carrier DNA置于沸水中煮 5min,立即置于冰上2min,如此重复一次;
(3)按照以下组份,分别配制转化体系:
|
实验组 |
阳性对照 | 阴性对照 |
|
4μg pBT3-bait 诱饵质粒 |
4μg pTSU2-APP 诱饵质粒 | 4μg pTSU2-APP 诱饵质粒 |
|
4μg pPR3-prey 猎物质粒 |
4μg pNubG-Fe65 猎物质粒 | 4μg pPR3-N 猎物质粒 |
|
100μL NMY51 酵母感受态细胞 |
100μL NMY51 酵母感受态细胞 | 100μL NMY51 酵母感受态细胞 |
|
10μL carrier DNA |
10μL carrier DNA | 10μL carrier DNA |
(4)缓慢轻柔加入500 μL PEG/LiAc,混匀颠倒;
(5)30℃水浴30 min,每10 min轻柔混匀一次;
(6)加入20 μL DMSO,轻柔混匀;
(7)42℃水浴15min,每5min轻柔混匀一次;
(8)800g离心1min,去上清,加入800 μL YPDA重悬,封口膜封上;
(9)30℃,150rpm,振荡培养1.5h;
(10)800g离心5min,弃上清,加入1mL 0.9% NaCl重悬,分别涂150μl于DDO、TDO/ 3-AT X mM(3-AT的浓度通过自激活验证获得)以及QDO平板;
(11)30 ℃倒置培养3-5 d,观察菌落数量及状态。
诱饵A蛋白与猎物蛋白共转化验证结果图
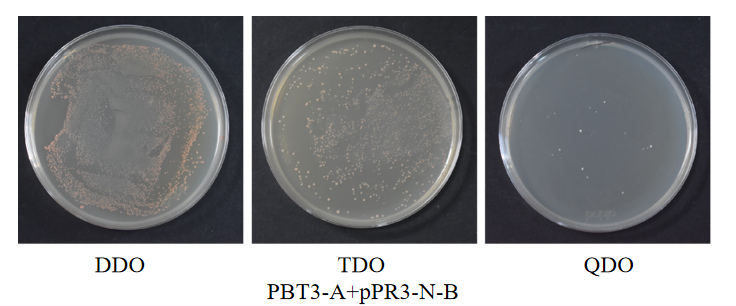
阴、阳对照组共转化验证结果图
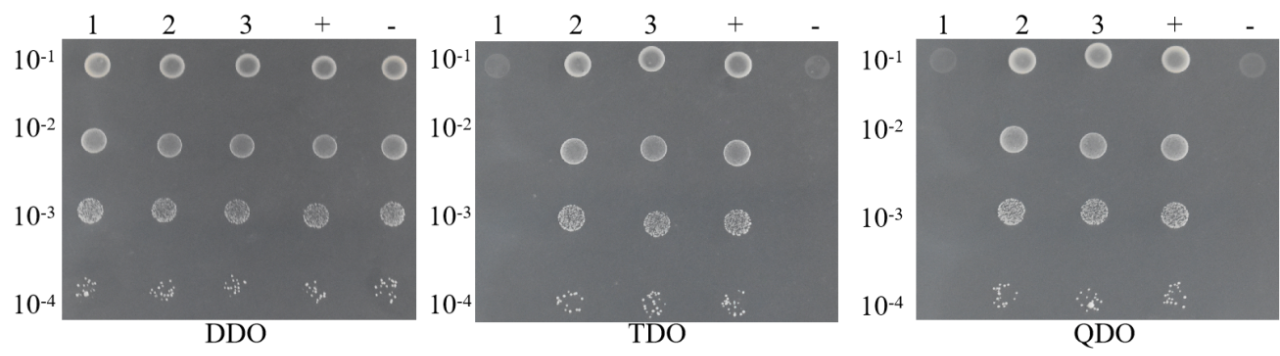
6.稀释点种
(1)依次从自激活、共转化实验组和共转化阴阳性对照组涂布的平板DDO上挑单菌落,分别接种于5mL DDO液体培养基,30 ℃ 250 rpm培养16 h;
(2)功能验证组、自激活组、实验组和阴阳对照组共5组,分别各取4个0.2ml PCR管,分别加入90μL 0.9% NaCl,依次编号为A、B、C、D;
(3)从1)摇好的菌液中取10 μL菌液加入管A中混匀,从混匀的A中取10 μL菌液加入管B中混匀,从混匀的B中取10 μL菌液加入管C中混匀,从混匀的C中取10 μL菌液加入管D中混匀;
从上述A、B、C、D管中各取10 μL菌液点种到平板DDO、TDO/ 3-AT X mM(3-AT的浓度通过自激活验证获得)、QDO上,30℃培养箱培养3-5d,观察菌斑生长情况。
稀释点种验证结果
三、常见问答
Q1.用筛出的阳性结果做点对点验证现阴性结果的原因是什么?
(1)酵母杂交本身也是存在假阳性的情况,有可能这两个基因根本不互作,合成全长进行验证自然也不会产生互作;
(2)对于双杂来说一般是两个结构域之间的互作,如果互作的两个基因的结构域恰好处于接触的状态,那么就可以发生相互作用;
(3)有些互作为瞬时互作,在文库筛选时可能捕捉到了互作,而在两个基因进行单独验证时就显示为阴性;
(4)有些互作为间接互作,可能需要依靠第三个基因的作用,促进这两个基因的互作;
(5)有些蛋白在天然情况下可能为膜蛋白或者是定位在膜上的蛋白,酵母杂交虽然是模拟体内互作,但是与天然状态下的蛋白还是有一定的差距的,为保证两个蛋白在同一空间,诱饵和猎物载体均带有核定位信号,会强行的将他们定位在核内,因此可能会产生假阳性的情况。
Q2.筛出的阳性结果为什么需要重新合成全长而不是片段做回转验证?
(1)片段的序列是基于测序结果得出来的,可能会有突变和缺失的情况,可能不是一个完整的结构域;
(2)将阳性测序结果进行NCBI数据库检索时,由于为片段式可能会匹配到很多的基因,没有办法确定为哪一个基因,需要进行试验确认才能定位到具体的基因,在进行后续的辅助实验EMSA,pulldown等实验时才有理论依据,否则在发文章时仅凭不能确定基因的片段并不能作为有力的支撑。
最新动态
-
04.30
Journal of Nanobiotechnology:苦瓜来源的外泌体样稳定p62表达以改善阿霉素心脏毒性
-
04.30
Theranostics-源自水果、蔬菜和草药的类外泌体纳米颗粒:治疗和药物递送的创新策略
-
04.22
Biology:藏红花花被片中外泌体样纳米颗粒的表征及其免疫刺激活性
-
04.22
酵母建库技术全解析:从原理到实战,助你轻松构建高质量文库
-
04.14
Int J Mol Sci | 植物性外泌体样纳米囊泡治疗早期骨关节炎的分离、表征和体外细胞研究
-
04.14
RNA pull down技术实验的应用场景
-
04.03
RIP技术如何破解RNA蛋白密码?
-
04.03
Food & Function:食用菌纳米囊泡突破-泌阳花菇提取物展现惊人辐射防护力
-
03.27
J. Adv. Res.|植物源性细胞外囊泡治疗IBD:多机制作用与药物载体潜力
-
03.27
植物外泌体:解锁中药未来的 “纳米密码”
 X
X 






