
科研人必须掌握构建载体的方法:超详细载体构建方案
一、什么是载体构建?
DNA需以质粒的形式才能稳定存在,那如何将DNA分子运送到受体细胞中去呢?这个时候我们就需要考虑能不能使用一种载体,既可以将其运送到受体细胞,又可以让其稳定存在。
实现这个想法就需要用到载体构建:将目的片段插入到线性化的载体骨架上,在酶的作用下,片段和骨架重新形成一个环形的质粒,再导入宿主细胞,实现目的基因在宿主细胞内的正确表达。
载体特点:
可自我复制:复制子 ori是一段具有特殊结构的DNA序列;
标记基因:有一个或多个便于检测的遗传表型,如抗药性、显色表型反应等;
酶切位点:有一个或几个限制性内切酶位点,便于外源基因片段的插入;
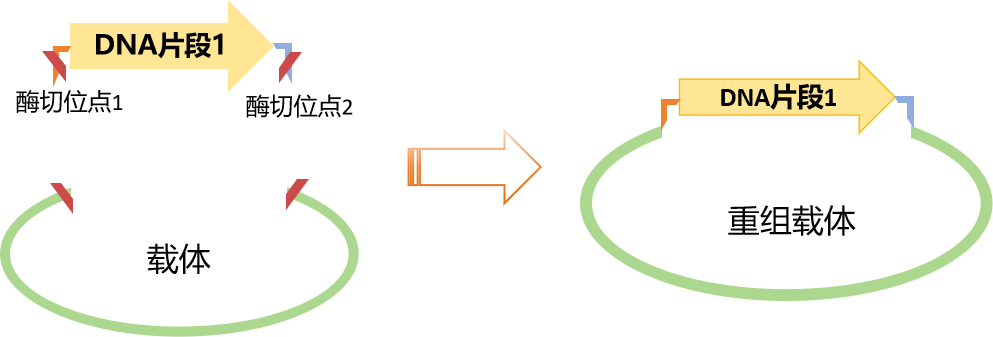
图1.载体构建原理
二、载体构建方案
1 、引物设计
♦ 可以使用NCBI的一个在线工具:
Primer-Blast网址:https://www.ncbi.nlm.nih.gov/tools/primer-blast/
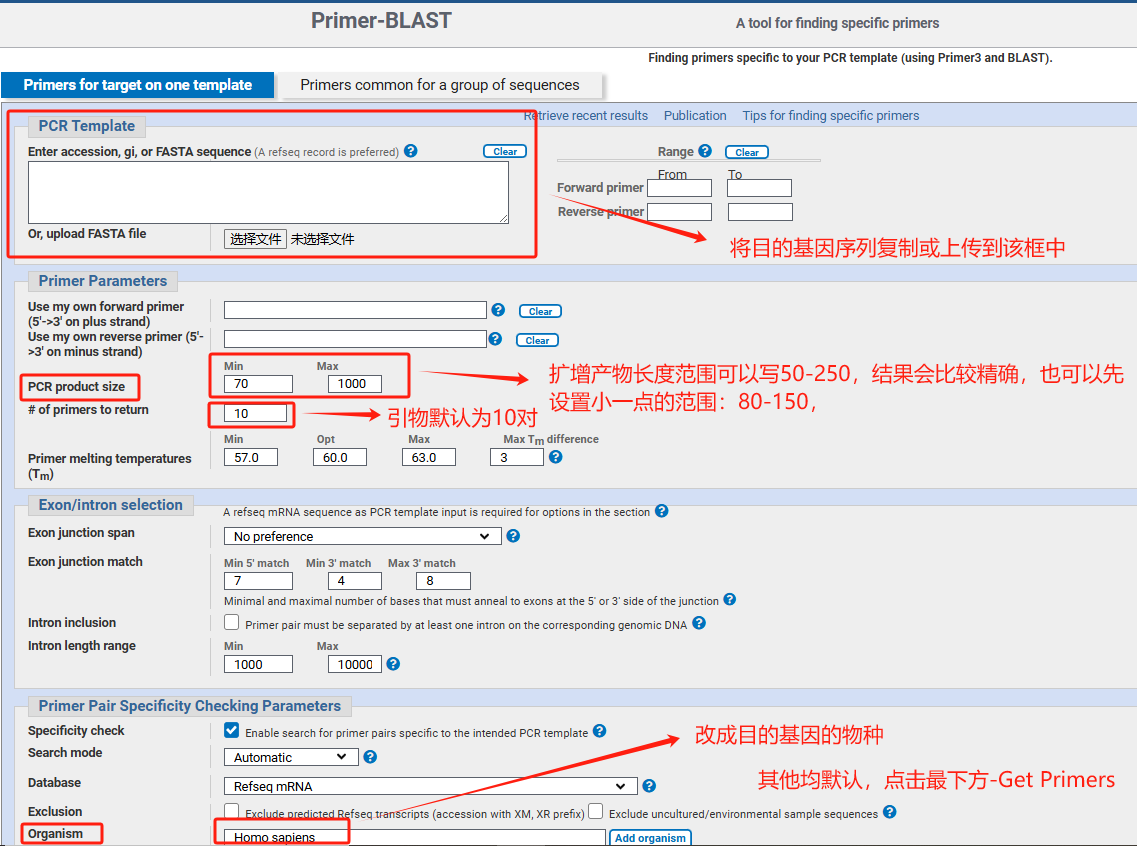
图2.Primer-Blast网站设计引物说明图
♦ 设计引物遵循规则
1、18-25bp长,引物GC含量40-60%;
2、引物Tm值58-65度,正反向引物相差小于4度,Tm=4℃(G + C)+ 2℃(A + T);
3、3’端最后5个碱基中的G或C少于2个,3端不要两个连续的G或C,引物3’端出现3个以上的连续碱基,如GGG 或CCC,也会使错误引发机率增加;
4、两引物间3端末相互配对小于2个碱基,一条引物内部5端和3端配对小于3个碱基,以免引物二聚体,一对引物间不应多于4个连续碱基的同源性或互补性;
5、3端最好不要T结尾,末位碱基为A 的错配效率明显高于其他3 个碱基,因此应当避免在引物的3’端使用碱基A,最好选择C或G;
6、引物内部4种碱基均匀分布,不要嘌呤(A、G)或嘧啶(T、C)连续存在,C、G或A、T不要过分集中,特别3端;
7、引物不要有发夹结构,引物自身连续互补碱基不能大于3bp(判断标准);
8、引物3′端不要终止于密码子的第3位。
2 、目的基因获得
(1)PCR扩增
设计引物:在扩增目的片段所用的正反引物中分别引入不同的两个酶切位点序列,而载体上也有这两个酶切位点(以EcoRI和KpnI为例);PCR扩增后用1%琼脂糖凝胶回收目的基因DNA条带,如果PCR条带非常单一,PCR结果特异性高,可不经凝胶电泳直接回收PCR产物
(2)在已有载体上截取(用相应的内切酶直接切下,电泳后回收)
(3)人工合成

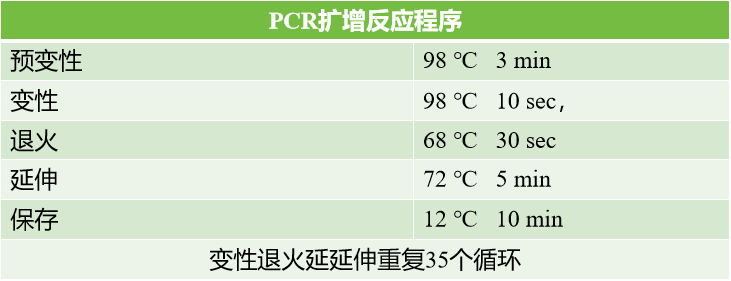
3 、目的片段和质粒的双酶切
限制性内切酶能特异地结合于一段被称为限制性酶识别序列的DNA序列之内或其附近的特异位点上,并切割双链DNA。
T4 DNA连接酶可选择性的将相同的黏性末端连接起来,从而确保目的片段以正确的方向接入载体。
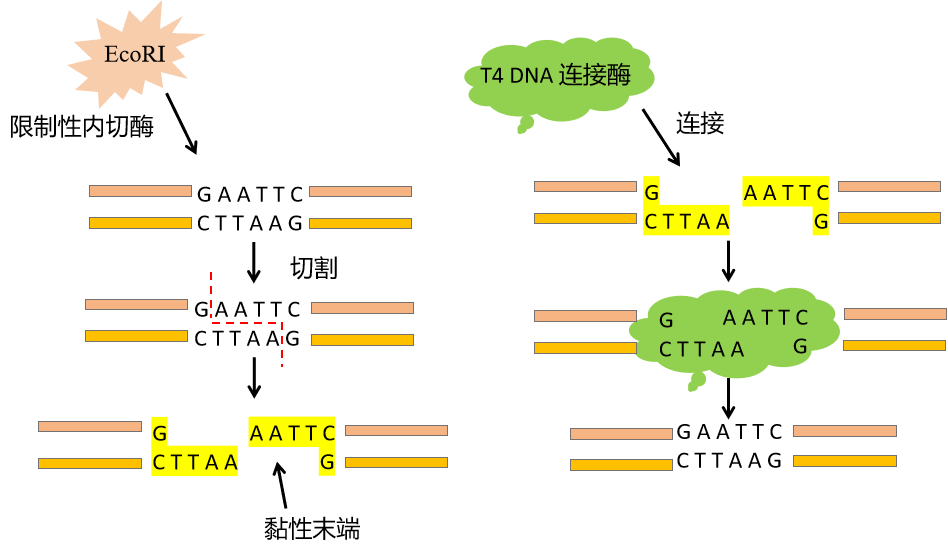
图3. 载体酶切和连接示意图
对载体及目的片段进行双酶切,用EcoRI和KpnI对质粒载体PUC57和目的基因PCR产物进行双酶切,酶切反应反应体系如下表所示(总体系 50 μL):
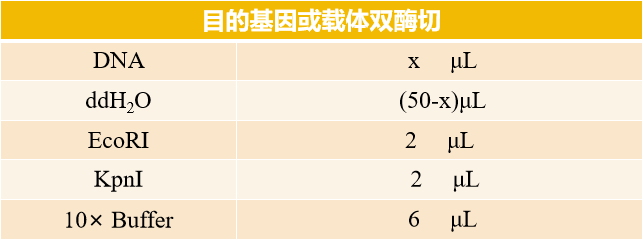
酶切体系配备好以后,37 ℃ 水浴(金属浴或 PCR 仪均可)2 h,对于双酶切后的载体和目的片段,需要跑胶后确认片段大小是否正确,将酶切好的条带切下进行胶回收即可(琼脂糖凝胶DNA回收试剂盒)。
注意:一般胶回收试剂盒回收率50%左右,如果DNA分子量大于10K后回收效率就更低,注意控制酶切体系中的DNA的量,量大酶切容易切不彻底,量少回收后浓度过低。
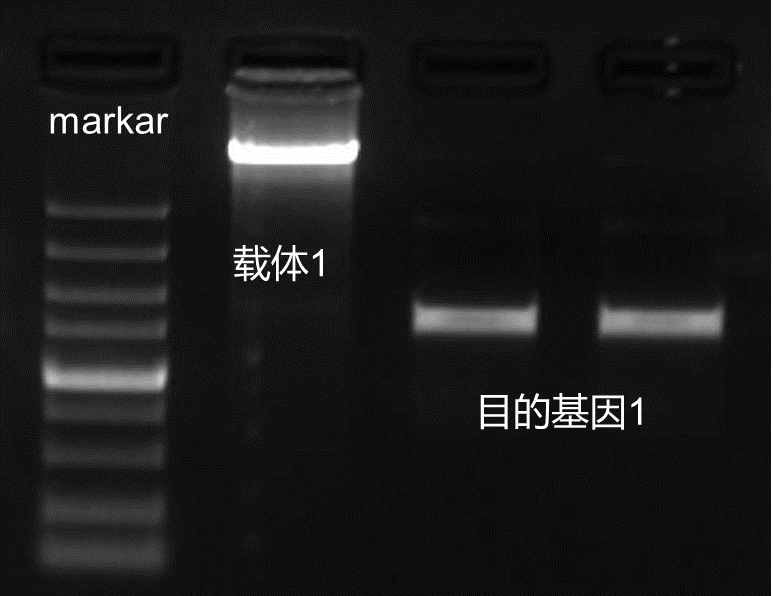
图4. 载体和目的基因双酶切胶图
4、连接转化
♦ 连接:质粒PUC57 EcoRI+KpnI酶切回收大片段与目的基因连接,在0.2 mL 微量离心管中制备连接反应液:1 µL 10×T4 DNA Ligase Buffer,约0.2 pmol 目的片段,约0.02 pmol 的载体骨架,0.5 µL T4 DNA Ligase,ddH2O up to10 µL,连接反应在16℃反应12小时。
♦ 转化:取10 µL连接产物与100µL DH5a感受态细菌混匀后冰浴30min,42℃热激90s,立即置冰上放置5min,加入预热至室温的700µL LB培养基,37℃恒温摇床培养50min,吸取 200µL的菌液,用移液器混匀后均匀涂布于含100µg/mL Amp抗性(根据实验要求选择抗生素)的LB平板上,37℃恒温培养箱倒置培养过夜。
注:目的片段的摩尔数应控制在载体摩尔数的3-10倍;摩尔数与质量:1 µg 1,000 bp DNA=1.52 pmol,1 µg pUC57 DNA(2,720 bp)=0.57 pmol。
5、挑取克隆提质粒验证
挑取5个单菌落接种于含5ml,100μg/mL Amp抗性的LB培养液中,300rpm,37℃恒温摇床培养过夜,对过夜的菌液进行扩增,选择阳性菌液,用质粒小量提取试剂盒提取质粒,再进行测序验证。
三、失败经验总结
♦ XbaI位点后面带TC:设计引物及选择酶切位点时注意避免。选择去甲基化的菌种如JM110,然后重新制备质。
♦ 紫外过度损伤粘性末端:使同一条DNA链上相邻的嘧啶以共价键连成二聚体,其中最容易形成的是TT二聚体导致DNA受到破坏。
♦ 片段加入越多越好?DNA加入过多,酶反应不完全。反应时间时间过长,酶的特异性降低。
♦ 材料试剂的保存与过程记录:实验记录和数据一定保存记录好,不然“一夜回到解放前”,纯纯白干喽!
最新动态
-
03.11
同为抗体,为什么一抗看“抗原种类”,二抗却看“一抗种属”?
-
03.11
J Nanobiotechnology(IF=12.6):蔓越莓“纳米邮差”递送麦角甾醇,重启早衰卵巢功能!
-
02.27
没有抗体?不是模式植物?DAP-seq照样搞定你的TF靶基因!
-
02.27
高分研究的共同选择:三篇权威文献背后的ChIP实验工具解析
-
02.27
表观遗传学进入“多维验证”时代,这些疾病机制再也藏不住了!
-
02.27
“药食同源”顶刊思路(IF=10.5)!来源鉴定+活性追踪+体内验证,全面解析无花果来源外泌体抑制乳腺癌骨转移的双重机制!
-
02.27
千年古方,纳米新生:科学家破解半夏如何“指挥”免疫细胞对抗肺癌!
-
01.29
技术跨界联用:表观遗传学进入“多维验证”时代,这些疾病机制再也藏不住了!文末有惊喜活动!
-
01.29
机制研究的金标准:金开瑞RIP/RNA pull-down试剂盒获多篇顶刊研究认可!
-
01.29
想测结合常数,但样本太珍贵?来试试MST技术
 X
X 






